卤代烷的碳正离子的结构与稳定性(Nat.Chem.Rh催化环丁烯的不对称交叉偶联构建环丁烷骨架)
含有环丁烷骨架的分子具有显著的生物学活性,广泛存在于许多天然产物和药物分子中,是有机合成中具有战略意义的中间体(图1a)。目前,构建手性环丁烷的方法主要有以下几种(Chem. Rev., 2003, 103, 1449):对映选择性环化反应、预先制备四元环的官能团化、多步环收缩和扩环策略等。虽然众多化学家在环丁烷骨架的构建方面做出了很多努力,但有关环丁烷的催化对映选择性合成却很少见,目前的方法主要分为两类——对映选择性闭环和预制备四元环的官能团化。具体而言,对映选择性闭环策略包括光催化和金属催化的[2 2]环加成反应(Science, 2013, 342, 840; Science, 2014, 344, 392; Science, 2018, 361, 68; Angew. Chem. Int. Ed., 2015, 54, 11918; J. Am. Chem. Soc., 2017, 139, 13628)以及特定底物和取代模式的环化策略(J. Am. Chem. Soc., 2015, 137, 10524; J. Am. Chem. Soc., 2017, 139, 10208)。另外,预制备的四元环可以通过导向基团控制的C-H键活化策略进行官能团化(J. Am. Chem. Soc., 2014, 136, 8138)以及活化环丁烯的加成反应(Angew. Chem. Int. Ed., 2013, 52, 6313; Angew. Chem. Int. Ed., 2020, 59, 2750)。
碳金属化(carbometallation)过程是不对称催化反应中最基本的过程之一,通常由环张力的释放触发,但对于未活化的环丁烯而言,这种过程却是未知的。相比之下,双键的加成反应可以由烯丙基位的吸电子基团触发。基于以上策略的启发,英国牛津大学化学系的Stephen P. Fletcher教授课题组成功地实现了铑催化不同种类环丁烯与芳基硼酸的碳金属化反应(图1b)。这些反应都有一个共同的中间体——环丁基铑配合物,后者是在对映选择性碳金属化步骤中形成的。对于不同的环丁烯,环丁基铑配合物可以通过C-H键插入或者链行走的方式生成不同类型的产物。例如,(1)A、E、F途径得到还原型Heck反应的产物;(2)B、C途径得到1,5-加成的产物;(3)D途径得到高烯丙基取代的产物。相关成果发表在Nature Chemistry 上。

图1. 催化不对称交叉偶联构建环丁烷骨架。图片来源:Nat. Chem.
如图1a所示,D4/5-HT2拮抗剂Belaperidone的骨架中含有 6-芳基-3-氮杂双环[3.2.0]庚烷,因此作者希望通过还原型Heck反应来构建该骨架。值得一提的是,原位生成的Rh/Segphos催化剂能够有效地实现芳基硼酸与环丁烯的还原型Heck反应(图2)。无论是间位还是对位取代的芳基硼酸(3a-3m)都能兼容该反应,并且可以耐受多种取代基,例如卤素(3d、3e、3g)、甲氧基(3b)、甲硫基(3c)、甲基(3f)和吡唑(3k),但是对位带有吸电子基团的硼酸(3h-3i)却收率较低。此外,邻位取代的芳基硼酸(3n)在标准条件下进行反应时获得了低对映选择性(52% e.e.),而将配体换为(S)-BINAP后便得到了较好的对映选择性(80% e.e.)。对于富电子的杂环硼酸(3p-3s),作者将配体换为(S)-DTBM-Segphos,也获得了良好的结果。当将环丁烯上的N-保护基换成Boc或Ts时,也能以良好的收率和优异的对映选择性得到相应的产物(3aa和3ba),而砜(3ca)或者环戊酮(3da)稠合的环丁烯也能实现这一转化。

图2. 环丁烯的氢芳基化反应。图片来源:Nat. Chem.
为了进一步探究反应机理,作者进行了一系列实验。如图3a所示,作者分离出了主要的副产物二环丁烷3a';另外,在重水中进行反应时得到了3a-C2-d。在此基础上,作者提出了一种可能的反应机理:1)PhB(OH)2和Rh-OH配合物I经转金属化得到中间体II;2)中间体II与环丁烯以anti的方式(相对于环丁烯上的取代基)进行Rh芳基化过程,从而以专一的立体选择性得到Rh-烷基配合物III;3)中间体III有两种反应途径。一方面,通过可逆的β-氢消除、1,2-氢迁移发生链行走过程,得到Rh-烷基配合物IV(图3b,链行走也可以通过1,3-氢迁移从VI到VIII);4)中间体IV与H2O发生质子化得到产物3并再生催化剂I。另一方面,中间体III中的Rh可以插入到芳基邻位的C-H键以产生Rh-芳基配合物V,后者与第二个环丁烯经Rh芳基化过程便可产生二聚体副产物3a'。

图3. 双环丁烯氢芳基化的应机理。图片来源:Nat. Chem.
出于对上述链行走过程的兴趣,作者设想能否对取代的环丁烷进行不对称1,5-加成。幸运的是,当使用 (S)-DTBM-Segphos为配体并在60 ℃下进行反应时,非共轭β,γ-不饱和羰基化合物4能与一系列芳基硼酸进行不对称1,5-加成反应(图4),并以优异的对映选择性和较好的产率得到相应的产物(5a-5j)。然而,邻位取代的芳基硼酸却不能实现这一转化。
同样地,作者进行了上述D2O实验,以88%的氘代率得到了5a-C1-d。虽然催化循环的前两个步骤很可能通过与底物1 相同的转金属化和碳金属化进行的(图3a),但是作者认为氧杂-π-烯丙基-Rh 配合物 XI通过β-氢消除/共轭1,4-氢加成形成(图5a),并且相对稳定的氧杂-π-烯丙基配合物可能会抑制竞争的二聚化途径。此外,作者在氧杂-π-烯丙基-Rh 配合物 XI 的质子化过程中观察到非对映选择性,说明环结合有利于cis-式构型。另一方面,cis-式二酯6在标准条件下进行反应时得到了产物7(图5b),主要分离出trans-trans非对映异构体,这表明氧杂-π-烯丙基-Rh 配合物的非对映选择性质子化能够远程立体控制。虽然环丁烯1和4是内消旋化合物,但是(±)-8的外消旋混合物在标准条件下进行反应时则得到了9和9'的混合物(~1:1,图5c)。最后,为了检验该方法的实用性,作者开发了两种高对映选择性方法来合成Belaperidone的中间体10,然后再进一步转化为Belaperidone(11,图5d)。该方法不仅催化剂用量低(2.5 mol%→0.5 mol%),而且还能以克级规模制备(5h,1.3 g)。

图4. 不对称1,5-加成反应。图片来源:Nat. Chem.
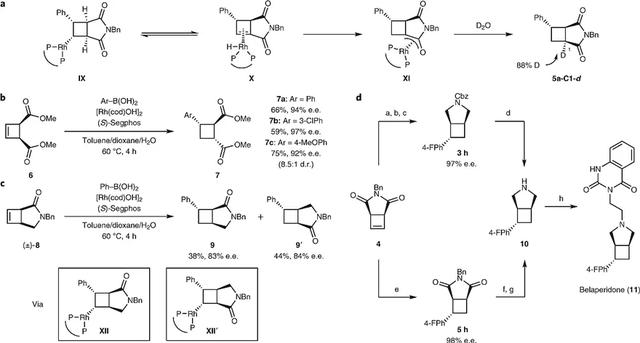
图 5. 金属化碳引发的不对称1,5加成反应。图片来源:Nat. Chem.
由于在非对称环丁烯的碳金属化中实现高区域选择性极具挑战性,因此作者选择螺环12为模型底物、非手性 Rh/dppf 为催化剂、CsOH为碱,在60 ℃下与一系列(杂-)芳基硼酸进行反应,实现了非手性产物(13a-13j)的区域选择性(图6)。此外,该方法还可以实现脂肪酸酰胺水解酶抑制剂候选药物 PF-048628531(14)的简洁、模块化合成(图7a)。与上述体系相反,氘代实验是通过 C-H 键插入途径发生在 C(2') 位。虽然通过碳金属化形成了不对称中间体,但在随后的 C-H键插入过程中失去了手性,从而得到了非手性产物(图7b)。为了进一步研究这种区域选择性的还原型 Heck 反应中的非对映选择性,作者将(±)-12a 置于标准条件下进行反应,以65%的收率和4:1的d.r. 值获得了13aa(图7c)。

图6. 区域选择性还原型Heck反应。图片来源:Nat. Chem.

图7. 还原型Heck反应和远端取代。图片来源:Nat. Chem.
在以上反应中,碳金属化后Rh-环丁基中间体的质子化是通过C-H 键插入或链行走过程完成的。受上述 1,5-加成过程的启发,作者尝试将这种加成策略用于取代反应。为此,作者将苯基硼酸加入到内消旋二乙酸酯 15 中,有效地在非共轭高烯丙基乙酸酯上发生了远程取代,得到带有两个立体中心和一个环外烯烃的产物 16a(图8)。如图7d所示,作者认为该反应是通过碳金属化、β-氢消除、氢化和 β-氧消除实现的。此外,该反应需要碱来实现高转化率,这可能是为了再生活性Rh-氢氧化物配合物I。值得一提的是,该反应能够耐受多种富电子芳基硼酸(16a-16j)以及其它离去基团(如二碳酸酯和二磷酸酯,16aa和16ba)。然而,对于缺电子芳基硼酸,则观察到还原型Heck反应为主要途径(17k-17m)。其中氢芳基化主要源于C-H键插入过程(对于缺电子硼酸来说更快)和随后的质子化过程(图7d),这种替代途径降低了远程取代产物(16e、16g、16h)的收率,并分离出大量的还原型Heck反应产物(17e、17g、17h)。需要指出的是,该过程的化学发散性取决于硼酸和配体的电性。例如,使用 (S)-DTBM-Segphos为配体,得到了氢芳基化产物 17n为主要产物,而将配体更改为 (S)-Segphos 则得到了远程取代产物 16n(图7e)。

图8. 远程取代。图片来源:Nat. Chem.
总结
Stephen P. Fletcher教授课题组在Rh催化的作用下,实现了环丁烯的不对称碳金属化过程,该过程不需要任何活化或导向基团,因此多种取代的环丁烯都可以进行这种转化。金属化的环丁烯可以遵循不同的途径(如C-H 键活化和链行走)以实现质子化或消除并再生催化剂,从而实现多种反应,如不寻常的 1,5-加成反应和非共轭烯烃上的高烯丙基芳基化反应。可以预见,该方法有望直接用于生物医学和农化研究以及精细化工行业中。
A catalytic asymmetric cross-coupling approach to the synthesis of cyclobutanes
F. Wieland Goetzke, Alexander M. L. Hell, Lucy van Dijk, Stephen P. Fletcher
Nat. Chem., 2021, DOI: 10.1038/s41557-021-00725-y
导师介绍
Stephen P. Fletcher
https://www.x-mol.com/university/faculty/2639
,免责声明:本文仅代表文章作者的个人观点,与本站无关。其原创性、真实性以及文中陈述文字和内容未经本站证实,对本文以及其中全部或者部分内容文字的真实性、完整性和原创性本站不作任何保证或承诺,请读者仅作参考,并自行核实相关内容。文章投诉邮箱:anhduc.ph@yahoo.com






