单萜的合成路线(复杂二萜的发散性全合成)
本文来自X-MOLNews
多环二萜化合物是一类结构复杂多样且具有重要生物活性的天然产物,其中在药用植物香茶菜中多见的ent-kauranes、ent-atisanes和ent-trachylobanes是与生物合成相关的二萜类天然产物家族(图1A)。这三个家族之间的主要结构差异在于C环和D环的碳环结构:ent-kauranes具有[3.2.1]双环体系,而ent-atisanes和ent-trachylobanes则分别具有[2.2.2]双环体系和[3.2.1.0]三环体系。尽管这些二萜具有引人注意的结构和生物活性,要想合成它们却颇具挑战,目前最常见的方法是从头合成。近年来,不少化学家也报道了相对不那么常见的半合成策略,例如Mander首次从赤霉素出发合成6,7-seco-ent-kauranes(Tetrahedron Lett., 1986, 27, 3927–3930),以及最近报道的从甜菊苷($ 0.65/g)出发合成atisane-型二萜生物碱(10,J. Am. Chem. Soc.,2014, 136, 12592–12595; J. Org. Chem., 2018, 83, 1606–1613;图1B)。甜菊苷价格廉价,是否也能用于制备高度氧化的ent-kauranes呢?事实上,这个合成过程并不容易。首先,甜菊醇(甜菊苷糖苷配基,9)的A、B或C环上缺少取代基,因此半合成修饰主要依赖于C19位,但是Hofmann-Löffler-Freytag或Suarez hypoiodite反应仅适用于C20甲基的修饰。其次,有关甜菊醇的远程C-H键氧化尚未报道过,这可能是由于环外烯烃C16-C17与C-H键氧化条件不兼容。
近日,美国斯克里普斯研究所(The Scripps Research Institute,TSRI)的Hans Renata教授课题组发展了一种化学酶促(chemoenzymatic)合成策略,即通过酶促氧化(远程生物催化羟基化)和化学氧化(导向C-H键氧化)相结合的混合氧化法来制备高度氧化的二萜化合物ent-kauranes、ent-atisanes和ent-trachylobanes(图1C)。具体而言,从ent-steviol出发,仅需最多10步便可合成9种复杂的天然产物。该方法不仅可以选择性地氧化先前方法无法修饰的位点,还可以使非生物骨架重排为其他基础结构。相关成果发表在Science 上,Xiao Zhang博士为第一作者。

图1. 复杂的二萜家族(ent-kauranes、ent-atisanes和ent-trachylobanes)。图片来源:Science
首先,作者对不同的氧化酶进行了筛选(图2)。先前的研究表明P450单加氧酶(PtmO5)能够催化ent-kauranol C11位的远程C-H键羟基化,但是必须要用单独的还原酶伴侣(reductase partner)来实现这一过程。然而,将CamA和CamB还原酶体系用于PtmO5中,发现其在全细胞和裂解液反应中的转化率非常低。经过一系列研究,作者发现将PtmO5与还原酶伴侣融合在一起或许是可行的方案。实验表明,所构建的嵌合蛋白PtmO5-RhFRed能够有效地促进9和13的C11位羟基化反应,从而选择性地氧化C环。此外,在单个大肠杆菌C41(DE3)菌株中共表达PtmO5-RhFRed、GroES和GroEL,可进一步提高9的C11位羟基化反应的收率(88%)。对于B环的选择性氧化而言,两种双加氧酶(Fe/aKGs)PtmO3和PtmO6均能催化C7位羟基化,但是PtmO6在大肠杆菌中表达后能更好地过量生产,并且能以较高的总催化转化数实现9(TTN = 1200)和13(TTN = 390)的C7位羟基化。鉴于P450BM3变体可用于萜烯骨架的选择性氧化,于是作者假设其中一些变体能够在9或13上进行类似的氧化。为了证实这一假设,作者在其酶库中初步筛选了P450BM3丙氨酸扫描变体,以检测9或13的羟基化情况,结果显示9没有实现羟基化,而13则形成了氧化产物。值得一提的是,变体BM3 MERO1 M177A能够选择性地生成C2位羟基化产物18,且没有任何过氧化或C16-C17环氧化副产物的生成。
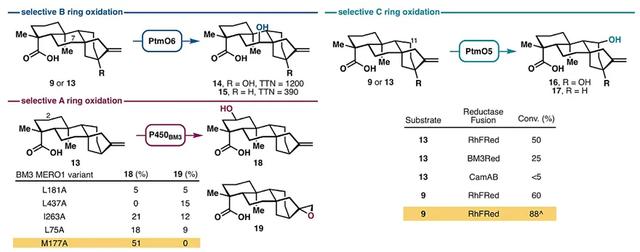
图2. 反应条件优化。图片来源:Science
接下来,作者通过发散性合成来制备ent-kauranes(图3):mitrekaurenone(21)、fujenoic酸(23)和pharboside糖苷配基(25)。具体而言,通过两步法(3°醇溴代和自由基脱卤)将甜菊醇(9)转化为ent-kaurenoic酸(13),后者经PtmO6氧化,以较好的收率得到单一的非对映异构体(15),并且该转化能够在表达PtmO6的大肠杆菌细胞的澄清裂解物中以克级规模进行。15经DMP氧化为相应的酮(20),后者在三溴化吡啶鎓的作用下进行α-氧化,同时引发分子内酯化,从而以5步实现了21的合成,总收率36%。对于23的合成而言,需要在B环上进行10电子氧化。起初,作者试图通过化学方法将20转化为α-羟基酮22,但是发现使用PtmO6能以较高的收率得到22,后者经NaIO4氧化(断裂C6-C7键)、DMP 氧化(iketaltal氧化),以7步、26%的总收率得到23。相比之下,25则是通过Burgess试剂将甲酯24的2°醇脱水成相应的烯烃,然后在OsO4和NMO的作用下,同时将两个烯烃转化为相应的syn-二醇单元,总共6步,总收率29%。

图3. PtmO6在化学酶促全合成中的应用。图片来源:Science
从甜菊醇9出发,合成rosthornin C(30)和rosthornin B(3)需要在C7位和C11位进行羟基化、C19位进行羧基还原,并在C15位引入羰基(图4)。那么,如何能够同时实现C7位和C11位的羟基化呢?作者发现酮26(由PtmO6羟基化和重铬酸吡啶鎓(PDC)氧化两步法制备而成)可以在PtmO5-RhFRed 的作用下于C11处进行区域选择性羟基化,尽管转化率中等。使用共表达的PtmO5-RhFRed和Opt13,能以65%的分离收率得到27,后者在Ac2O和DMAP的作用下,选择性地在C11位进行乙酰化。接下来,需要将C19位羧酸还原为醇29,即先将酸转化为相应的酰基咪唑,然后用NaBH4处理,但是该过程会伴随着将C7位酮还原为α-醇。29在SeO2和IBX的作用下选择性地氧化为酮,从而将烯酮单元安装在D环上,以总共七步、12%的总收率完成了30的合成。最后,对C19位伯醇进行选择性乙酰化便可得到3。
鉴于Fischericin B(2)含有一个笼状醚基,因此作者从9出发,经PtmO5氧化得到C11位羟基化产物,后者在强酸性条件(TFA)下得到所需的笼状醚基31。接着将31的游离酸甲基化,然后对其C13位叔醇进行Barton脱氧反应、C19位酯基进行还原得到34,后者在Suarez hypoiodite反应条件下得到碘醛35,该碘醛可进一步被氧化并进行分子内关环反应,从而以9步(总收率:25%)实现了2的全合成。值得一提的是,在2的全合成过程中利用了混合氧化法,即酶促氧化(C11位羟基化)和化学氧化(C20位C-H键氧化)。

图4. PtmO5-RhFRED在化学酶促全合成中的应用。图片来源:Science
由于甜菊苷可一步转化为异甜菊醇(36),因此作者尝试合成ent-atisane和 ent-trachylobane(图5A),但是该过程需要在C12位安装能后续生成碳正离子的基团。前文曾提到,PtmO5-RhFRed能够有效地促进C11位羟基化反应,因此作者设想36在PtmO5-RhFRed的作用下进行C11位羟基化后,将所得的醇从C11位迁移到C12位,事实上,36与PtmO5-RhFRed反应竟意外地得到C12位羟基化产物37,收率92%,且能以克级规模进行。37在三氟甲磺酸(TfOH)作用下,发生预期的Wagner-Meerwein重排得到38,后者经L-selectride还原得到醇39。接着,39在BF3•Et2O和Et3SiH的作用下进行还原性重排,经3次回收以61%的收率得到ent-trachylobane 40。作者推测这种重排可能是通过C13醇离子化进行的,随后形成非经典碳正离子并在C15位进行选择性还原淬灭。
先前的研究表明38与天然ent-atiserenoic酸前体的结构高度相似,是一种有用的中间体,可通过酶促羟基化反应获得更多的氧化ent-atisanes。如图5B所示,它可以在PtmO6的作用下将C7位氧化为醇41,后者经Wolff-Kishner脱氧(C13位酮)和PDC氧化(C7位-OH)得到酮42(spiramilactone C(5)的中间体)。或者,也可以在BM3 MERO1 M177A的作用下将C2位氧化为醇43,且未观察到C15-C16烯烃的环氧化。43经Wolff-Kishner还原得到44,后者可进一步转化为二萜类生物碱cochleareine(6)。

图5. 通过非生物骨架重排和混合氧化法合成复杂的天然产物。图片来源:Science
如图5C所示,中间体40可发散性合成mitrephorones A、B、C(7、49、8)。具体而言,在BM3 MERO1 M177A的作用下,将其C2位进行氧化。在较高酶底物比(enzyme-to-substrate ratio)的情况下,该过程还能进行迭代氧化,从而在C2位安装酮。接着经PtmO6氧化、PDC氧化得到二酮45,后者经PtmO6氧化于C6位安装OH。由于中间体46不稳定,需要将C19位酸进行快速甲基化并在C6位进一步氧化,然后进行酮-烯醇互变异构,便完成了mitrephorone C(8)的合成。此外,用PtmO6对中间体40的C7位进行氧化便可得到47。那么,接下来的问题在于如何在C6-C7上安装二酮单元呢?起初,作者尝试将C7-OH脱水成相应的烯烃,然后用钌催化剂直接氧化来安装C6-C7二酮单元。然而,该方法不仅收率低,而且会伴有副产物的生成。于是,作者另辟蹊径,将47氧化为相应的酮48,后者在PtmO6的作用下进行C6位羟基化、甲基化并用PDC氧化,便可实现mitrephorones B(49)的合成。值得一提的是,作者偶然发现49能够在空气下缓慢氧化形成7(7 days后收率:45%;14 days后收率:65%)。
总结
Hans Renata教授课题组发展了一种化学酶促合成策略,仅需最多10步便可实现9种复杂二萜化合物的全合成。该策略的关键在于细菌来源酶的“强迫营业”——催化它们在自然界不曾催化的反应。三种不同的酶,对A、B、C环中特定位点进行羟基化反应。并且,该过程还能和化学C-H键氧化相结合,弥补了纯化学方法的不足。该反应不仅开辟了复杂二萜化合物全合成的新思路,而且为化学催化与生物催化相结合奠定了实质性基础。
Divergent synthesis of complex diterpenes through a hybrid oxidative approach
Xiao Zhang, Emma King-Smith, Liao-Bin Dong, Li-Cheng Yang, Jeffrey D. Rudolf, Ben Shen, Hans Renata
Science, 2020, 369, 799-806, DOI: 10.1126/science.abb8271
,免责声明:本文仅代表文章作者的个人观点,与本站无关。其原创性、真实性以及文中陈述文字和内容未经本站证实,对本文以及其中全部或者部分内容文字的真实性、完整性和原创性本站不作任何保证或承诺,请读者仅作参考,并自行核实相关内容。文章投诉邮箱:anhduc.ph@yahoo.com






