阿尔茨海默病循证防治(ATAD3A-新的阿尔茨海默病治疗靶点)
导读:
阿尔茨海默病 (AD) 是最常见的年龄依赖性神经退行性疾病。其病因不明。AD的特点是淀粉样蛋白沉积、神经原纤维缠结、突触丧失和进行性认知衰退。研究发现,脂质稳态的扰动是 AD 的另一个特征。阿尔茨海默病 (AD) 的易感性可能源于脂质代谢紊乱,然而,其潜在的机制仍然难以捉摸。近期《Nature Communications》期刊发表“ATAD3A oligomerization promotes neuropathology and cognitive deficits in Alzheimer's disease models”的研究论文。作者将含有 ATP 酶家族 AAA 结构域的蛋白质 3A (ATAD3A)(一种线粒体 AAA-ATP 酶)作为胆固醇代谢障碍与 AD 表型联系起来的分子开关,发现在 AD 的神经元模型、5XFAD 小鼠模型和死后的 AD 大脑中,ATAD3A在线粒体相关内质网膜(MAMs)寡聚和积累,并通过抑制CYP46A1基因表达诱导胆固醇积累。ATAD3A 和 CYP46A1 合作促进 APP 加工和突触丢失。通过杂合 ATAD3A 敲除或 DA1 药理抑制 ATAD3A 寡聚化可恢复神经元 CYP46A1 水平,使脑胆固醇转换和 MAM 完整性正常化,抑制 APP 加工和突触损失,从而减少 AD 转基因小鼠的 AD 神经病理改变和认知缺陷。这些发现揭示了 ATAD3A 寡聚化在 AD 发病机制中的作用,提示 ATAD3A 作为 AD 的治疗靶点的潜力。

AD模型中的 ATAD3A 寡聚化增加
作者首先通过对总共10,072 个优先疾病表型和23,499个优先基因进行虚拟筛选,对ATAD3A在AD表型、基因和途径中的优先级进行了计算分析。数据挖掘显示,ATAD3A与AD特异性表型和AD相关基因密切相关,分别位于前20.82%和14.49%(p<0.0001;补充图1a-c )。ATAD3A 排名靠前的途径与蛋白质代谢、基因转录、免疫反应和神经变性有关(补充图 1d)。这些数据表明 ATAD3A 可能参与AD病理学的发展。
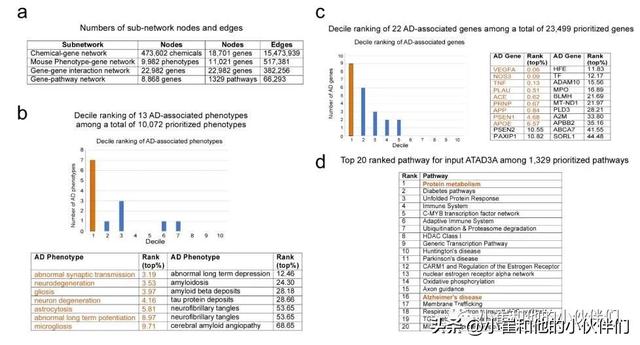
补充图1 计算分析以确定ATAD3A和AD之间的关系
为了确定AD中ATAD3A的变化,作者首先评估了各种AD实验模型中的 ATAD3A 寡聚化。在非还原条件下(即缺乏β-巯基乙醇,β-ME),以时间和剂量依赖的方式暴露于低聚aβ1-42肽的永生小鼠海马HT-22神经元和Neuro2a神经母细胞瘤细胞中,以及毒性Aβ处理的小鼠初级皮层神经元中(图1b),ATAD3A低聚物水平增加(图1a,补充图2a)。同时,作者证实在稳定的 APP 野生型(APP[wt])和 APP 瑞典突变体 (APP[swe])表达的 Neuro2a 细胞在化学交联剂 (双马来酰亚胺-己烷, BMH) 存在下, ATAD3A 寡聚体表达增加,在 APP[swe]表达细胞中观察到寡聚体表达更加活跃(图1c)。同样地,在非还原条件下,来自AD患者死后海马的总蛋白质裂解物中的ATAD3A寡聚体增加(图1d)。ATAD3A 寡聚体在 5XFAD AD 小鼠的皮质、海马和丘脑中也升高,但在其他脑区没有升高(图 1e,补充图 2c),与区域特异性Aβ聚集和人类APP表达相一致。相对于野生型(WT)小鼠,5XFAD 小鼠皮质中的 ATAD3A ATP 酶活性没有变化(补充图 2d)。ATAD3A ATP酶死亡突变体 ATAD3A-K358E-Flag 的过表达对 ATAD3A 寡聚化也没有影响(补充图 2e)。因此,ATAD3A 寡聚体的形成与蛋白质的酶活性无关。

图1 AD模型中的异常ATAD3A寡聚化
免疫组织化学分析显示,AD患者死后海马中的 ATAD3A 染色高于正常受试者(图 1f,补充图 2g)。此外,与正常受试者相比,作者观察到 AD 患者死后皮层中抗 NeuN 抗体免疫阳性的神经元中 ATAD3A 免疫密度增加(图 1g)。在 3 个月大的 5XFAD AD 小鼠大脑的皮质层 IV-V、皮层下和海马区中始终观察到 NeuN 免疫阳性细胞中 ATAD3A 免疫密度的增加(图 1h,i,补充图 2h)。此外,ATAD3A在AD患者和小鼠死后皮层的 APP 免疫阳性细胞中富集(图 1j)。ATAD3A 的 mRNA 和总蛋白水平在 3 个月大的 WT 和 5XFAD 小鼠大脑中相当(补充图 2i,j)。因此,AD患者和小鼠大脑中 ATAD3A 的免疫密度升高可能是由于ATAD3A 寡聚化增加,这与我们之前的观察结果一致。总的来说,作者的数据表明在 AD 表现期间 ATAD3A 寡聚化异常增加,这支持了我们的计算分析结果。

补充图2 异常 ATAD3A 寡聚化与 AD 有关
ATAD3A在MAM处积累并损害AD模型中的MAM完整性
作者团队和其他人先前发现ATAD3A定位于线粒体相关的接触位点,并且可能在 MAM 中富集。小鼠大脑的线粒体亚隔室分离揭示了MAM组分中的ATAD3A富集。ATAD3A存在于与VDAC和SigmaR1相同的线粒体部分中,这两种蛋白质已定位于MAM(补充图 3a)。值得注意的是,与WT小鼠相比,5XFAD 小鼠中ATAD3A在MAM部分的分布显著增强(补充图 3a)。用寡聚Aβ42处理的Neuro2a细胞的MAM部分的ATAD3A也显著增加(补充图3a)。ER和线粒体之间的生理接触距离在10到 30 nm之间,这允许使用原位邻近连接测定 (PLA) 来评估 ER-线粒体束缚和蛋白质在MAMs上的定位。在使用抗sigmar1和抗vdac抗体对脑组织切片进行染色后,作者观察到5XFAD小鼠皮层(图 2a)和AD患者死后皮层(图 2b)中PLA阳性点状突起的数量是对照样本的两倍。这些5XFAD小鼠和AD患者大脑中PLA阳性泪点的大小也大于对照组(图 2a,b)。此外,高分辨率显微镜显示在暴露于低聚aβ1-42肽的HT-22细胞中,IP3R3和VDAC之间有更高的共定位(补充图 3b)。这些数据表明AD模型中的MAM栓系增强,这与 AD[10][35] 中 MAM 的超连通性一致。在用抗 ATAD3A 和抗 FACL4(MAM标记)抗体染色后,作者观察到5XFAD小鼠和AD患者死后皮层中 PLA 阳性斑点的数量增加了大约两倍(图 2a,b)。与对照组相比,5XFAD AD 小鼠和AD患者死后大脑中ATAD3A和FACL4之间的PLA阳性斑点的大小也显著增加(图 2a,b)。这些结果也证明了 AD 患者和小鼠大脑中 MAM 处的 ATAD3A 积累。类似地,在HT-22细胞中,当细胞用抗ATAD3A和抗FACL4 抗体染色时,用寡聚Aβ1-42 肽处理会增加 PLA 阳性斑点的数量和大小(图 2c)。在对 ATAD3A 和细胞色素 c(一种线粒体膜间隙蛋白)、IP3R3(一种MAM蛋白)或SigmaR1和mtCO1(一种线粒体内膜蛋白)或FACL4和mtCO2 (一种线粒体内膜蛋白)染色的HT-22细胞中未观察到PLA阳性信号(补充图3c),表明与ATAD3A和FACL4相关的PLA阳性斑点的特异性。

补充图3 ATAD3A 出现在 MAM 中
基于在各种AD模型中观察到的ATAD3A寡聚和在MAM处的积累,作者确定了异常ATAD3A寡聚对终末软骨栓系的影响,终末软骨栓系是MAM完整性和活性的标志物。作者使用慢病毒ATAD3A shRNA敲除HT-22细胞中的ATAD3A,或用他们开发的DA1肽处理细胞以阻断ATAD3A寡聚化。在存在寡聚Aβ1-42肽的情况下,与对照组相比,ATAD3A下调或DA1处理显著减少了用抗IP3R3和抗VDAC抗体或抗SigmaR1和抗VDAC染色的Aβ处理的HT-22细胞中PLA阳性斑点的数量(图2d,补充图3d)。DA1处理也减少了Aβ诱导的线粒体片段化(补充图3e)。与之前的研究一致,寡聚Aβ42增加了IP3R3和FACL4蛋白水平,这被ATAD3A敲除所消除(图2e)。
作者之前证明了去掉前50个氨基酸的截断ATAD3A突变体(ATAD3A ΔN50-Flag)增强了ATAD3A的寡聚化。在这里,作者用ATAD3A-wt-flag或截断突变体ATAD3A-ΔN50-Flag转导HT-22细胞,并评估MAM栓系。当HT-22细胞用抗sigmar1和抗VDAC抗体或抗IP3R3和抗VDAC抗体染色时,过表达ATAD3A-wt- flag和ATAD3A-ΔN50-Flag显著增加了PLA阳性点状体的数量,其中表达ATAD3A-ΔN50-Flag突变体的细胞中pla阳性点状体的数量更多(图2f)。ATAD3A-wt-flag和ATAD3A-ΔN50-Flag的表达没有改变MAM相关蛋白的水平(补充图3f)或ATAD3A ATP酶活性(补充图2f)。总的来说,结果表明,ATAD3A 寡聚化和 MAM 处的积累可以诱导 ER-线粒体束缚的超连接,与AD样病理学有类似。

图2 在AD条件下,ATAD3A寡聚会损害MAM的完整性
ATAD3A单倍体不足可减少5XFAD 小鼠的认知缺陷和AD病理
接下来,作者确定了抑制异常 ATAD3A寡聚化是否会影响小鼠的AD相关神经病理学和行为缺陷。ATAD3A纯合子敲除导致小鼠胚胎致死,但杂合子敲除小鼠是正常且可生育的。作者通过表达CMV重组酶,在所有组织中敲除ATAD3Afl/fl小鼠的一个ATAD3A等位基因(以下简称CMV;ATAD3Afl/ )。然后生成了双突变体5XFADhet;通过5XFAD杂合子小鼠与CMV杂交获得ATAD3Afl/ 小鼠(补充图 4a)。CMV;ATAD3Afl/ 小鼠和5XFADhet;CMV;ATAD3Afl/ 双突变小鼠以预期的孟德尔比率出生,与WT和5XFAD同窝小鼠无法区分,表明缺乏明显的发育缺陷。在四种基因型之间没有观察到体重的显著差异(补充图4b)。Western印迹分析显示CMV的皮质和海马中总ATAD3A蛋白水平较低;ATAD3Afl/ 和 5XFADhet;CMV;ATA-D3Afl/ 小鼠比WT 同窝小鼠和 5XFADhet;ATAD3A / 小鼠低(图 3a)。线粒体亚区室的蛋白质水平(外膜蛋白VDAC和Tom20;ClpP,基质蛋白;ATPB,内膜蛋白)在所有四种小鼠基因型中相当(图 3a),表明杂合敲除ATAD3A 没有改变线粒体质量。此外,免疫荧光染色证实CMV中ATAD3A下调;ATAD3Afl/ 和5XFADhet;CMV;ATAD3Afl/ 小鼠(补充图 4a)。这些数据证实了ATAD3A基因敲除小鼠的特异性杂合性。与作者的发现一致,ATAD3A 寡聚体在3个月大的 5XFADhet;ATAD3A / 小鼠显著高于年龄匹配的 WT 同窝小鼠。值得注意的是,年龄匹配的5XFADhet中ATAD3A 寡聚体的水平;CMV;ATAD3Afl/ 小鼠恢复到在 WT 同窝小鼠中观察到的水平(图 3b)。因此,去除ATAD3A基因的一个拷贝会减少AD小鼠中的 ATAD3A 寡聚化。
为了评估 5XFADhet;CM-V;ATAD3Afl/ 小鼠的空间学习和记忆,作者对所有四种基因型的小鼠进行了Y迷宫和Barnes迷宫测试。通过Y迷宫测试,5XFADhet;ATAD3A / 小鼠在6个月大时其短期认知能力下降。相比之下,年龄匹配的 5XFADhet;CMV;ATAD3Afl/ 小鼠在Y迷宫测试中具有改善的自发改变率,达到与 WT小鼠观察到的水平相似的水平(图 3c)。在巴恩斯迷宫测试的第5天和第12天评估中,与WT和CMV相比,8个月大的 5XFADhet;ATAD3A / 小鼠花费了更长的时间和犯更多的错误才能找到目标逃生箱;ATAD3Afl/ 小鼠,表明认知能力下降。年龄匹配的 5XFADhet;CMV;ATAD3Afl/ 小鼠在第 5 天的潜伏期和错误数量显著减少,并且在第12天认知活动仍然得到改善(图 3d;补充图 4c)。与之前的报告一致,5XFAD小鼠在旷场测试期间过度活跃。5XFADhet;CMV;ATAD3Afl/ 小鼠在旷场测试中显示出与 WT 小鼠相似的归一化总距离(补充图 4d)。这些数据表明减少的ATAD3A寡聚化改善了5XFAD AD小鼠的空间学习和长期记忆。
作者用抗SigmaR1和抗VDAC或抗ATAD3A和抗FACL4抗体对四种基因型小鼠的脑切片进行染色,然后进行PLA以评估MAM在体内的完整性。作者观察到8个月大的5XFADhet;ATAD3A / 皮质中PLA阳性斑点的数量增加,在年龄匹配的 5XFADhet;CMV;ATAD3Afl/ 小鼠中降低到与在WT小鼠中观察到的水平相似的水平(图 3e,补充图 4e),通过ATAD3A杂合子敲除证明了MAM超连接性的正常化。为了评估5XFAD AD小鼠中的淀粉样蛋白聚集,作者用标记淀粉样蛋白聚集的抗6E10抗体对代表四种基因型的8个月大小鼠的脑切片进行染色。来自5XFADhet;ATAD3A / 小鼠的 CA1 和海马下托区域和皮质中6E10 淀粉样蛋白沉积的数量增加,淀粉样蛋白斑覆盖的面积更大。这种异常的淀粉样蛋白积累在年龄匹配的5XFADhet;CMV;ATAD3Afl/ 小鼠中减少(图 3f)。神经炎症是AD的另一个病理标志。作者发现,与5XFADhet;ATAD3A / 小鼠相比,5XFADhet;CMV;ATAD3Afl/ 小鼠皮质中Iba1(小胶质细胞标记物)和GFAP(星形胶质细胞标记物)的免疫密度显著降低(图3g),表明AD相关胶质细胞增生减少。这些数据表明,在5XFAD AD小鼠中,增强的ATAD3A寡聚基因的减少可降低神经病理。

图3 ATAD3A 杂合子敲除对 5XFAD 小鼠具有神经保护作用
DA1对ATAD3A寡聚化的抑制在AD模型中具有神经保护作用
作者接着开发了一种基于多肽的ATAD3A抑制剂DA1,它在HD的实验模型中特异性结合ATAD3A蛋白并在压力条件下抑制其寡聚化。DA可以通过小鼠的血脑屏障,并且在长期治疗期间被小鼠耐受(补充图 5a-d)。此外,在渗透泵植入后一天,FITC缀合的DA1荧光信号在WT小鼠的大脑中显著积累(补充图 5e),证实了皮下注射DA1给药可以进入小鼠大脑。在暴露于寡聚Aβ1-42 肽的培养的HT-22细胞中,DA1肽消除了ATAD3A寡聚化和升高的免疫密度信号(补充图 6a)。为了测试DA1肽的体内功效,作者用DA1或对照肽皮下处理纯合5XFAD(5XFADhomo) 从1.5到9个月大的小鼠使用Alzet微型泵TAT(1 mg/kg/天)(补充图 6b)。与5XFADhet相比,5XFADhomo小鼠发生淀粉样病变的速度更快,同时具有更广泛的神经表型,并且缺乏基因剂量效应,因此更适合评估DA1治疗的疗效。用DA1治疗不仅消除了ATAD3A寡聚的异常增加,而且还降低了5XFADhomo小鼠中增强的ATAD3A免疫敏感性(图4a,补充图6a),证实了DA1在体内的抑制作用。此外,用DA1治疗可抑制AD小鼠的MAM超连接,如用抗SigmaR1和抗VDAC或抗ATAD3A和抗FACL4抗体染色后6个月大5XFAD AD小鼠大脑中PLA阳性斑点数量减少(图4b,补充图6c)。值得注意的是,与使用对照肽治疗的年龄匹配的5XFAD小鼠相比,持续DA1治疗显著改善了5XFAD小鼠在6个月大时的Y迷宫试验(图4c)和8个月大时的巴恩斯迷宫试验(图4d;补充图6d)中的表现。此外,DA1治疗增强了8.5个月大5XFADhomo小鼠的筑巢能力,该方法被用作补充行为测定,因为它对AD的空间记忆和海马神经元损伤敏感(补充图6e)。DA1治疗还使6个月大5XFADhomo小鼠相对于WT小鼠的总旅行距离正常化(补充图6f)。经TAT和DA1处理的WT或5XFAD小鼠的体重相当(补充图6g)。重要的是,DA1持续治疗(七个月)对WT小鼠的行为状态或体重没有明显影响(图4,补充图6),表明缺乏长期毒性。
免疫组化显示用DA1治疗降低了6个月大的5XFADhomo小鼠皮质中的Aβ 覆盖面积和Aβ免疫密度(图 4e)。治疗还消除了5XFADhomo小鼠的皮质和海马中的Iba1 和GFAP 免疫反应性(图 4f,补充图 6h),表明抑制了神经炎症。在5XFADhomoAD小鼠的海马体和皮质V层中观察到神经元丢失,同时伴有淀粉样蛋白聚集和神经炎症。我们对6个月大的5XFADhomo小鼠的脑组织进行了荧光染色,荧光探针可以选择性结合退化神经元和抗Neun抗体。我们观察到海马和皮层CA1区NeuN 细胞中FJC阳性荧光信号,表明神经元正在丢失(图4g)。DA1治疗 5XFADhomo小鼠可降低神经元退变程度。事实上,与使用对照肽TAT处理的5XFADhomo小鼠相比,FJC荧光信号密度降低了70%以上(图4g)。此外,持续的DA1治疗没有引起WT小鼠的神经炎症和神经退行性反应(图4,补充图6)。因此,DA1抑制ATAD3A寡聚降低了5XFAD小鼠AD的神经病理学和认知功能缺损,这与作者在杂合ATAD3A敲除5XFAD小鼠中所做的观察结果一致(图 3;补充图 4)。

图4 DA1抑制ATAD3A寡聚化对AD小鼠具有神经保护作用
异常的ATAD3A寡聚化抑制AD模型中CYP46A1介导的脑胆固醇转换
为了研究ATAD3A寡聚化介导AD相关神经病理学的机制,作者对 5XFADhet;CMV;ATAD3Afl/ 小鼠的脑组织进行了公正的无标记蛋白质组学分析。作者从3个月大的小鼠身上采集了大脑皮层,此时小鼠的ATAD3A寡聚化增强,但没有表现出明显的淀粉样蛋白积累或认知缺陷。在 5XFADhet;CMV;ATAD3Afl/ 的皮质组织中鉴定的2639种蛋白质中,作者专注于在5XFADhet;ATAD3A / 中改变的蛋白质](即,相对于WT小鼠>1.5倍下调或上调)并同时通过ATAD3A的杂合敲除进行修饰(即,相对于5XFADhet上调或下调>1.5倍;ATAD3A / ;图 5a)。随后将总共 774 种符合这些标准的蛋白质(图 5,标有 *)用于通路富集分析。KEGG分析的图形比较显示,“代谢途径”和“阿尔茨海默氏病途径”类别位居前两个蛋白质富集途径,其中“脂质代谢过程”和“胆固醇代谢过程”富集的蛋白质分别受到的影响最大(图 5a,补充图7a)。随后的GO生物途径分析显示CYP46A1在“脂质代谢过程”和“胆固醇代谢过程”之间重叠(图5a,补充图7a)。此外,CYP46A1在AD小鼠中被评为杂合ATAD3A敲除调节的最佳候选药物。特别是,CYP46A1在5XFADhet;ATAD3A / 小鼠中相对于WT小鼠下调,并在 5XFADhet;CMV;ATAD3Afl/ 小鼠中恢复(图 5a,补充图 7a)。CYP46A1是一种大脑特异性酶,可催化胆固醇24-羟基化,这是从大脑中去除胆固醇的主要机制。作者假设ATAD3A寡聚化可能通过抑制CYP46A1介导的脑胆固醇代谢导致 AD相关的神经病理学和认知缺陷。
CYP46A1免疫密度在5XFAD小鼠大脑的皮质、海马CA1和下托中降低,主要在 NeuN[ ] 神经元细胞中(图 5b-e,补充图 7b),这与之前的发现一致[44]。相反,NeuN 神经元中CYP46A1染色的强度为在用DA1肽处理的 5XFADhet;CMV;ATAD3Afl/ 小鼠和5XFADhomo小鼠中显著升高(图 5b-e,补充图7b)。通过ATAD3A的杂合敲除也恢复了5XFADhet小鼠大脑中降低的CYP46A1蛋白质水平(补充图 7b)。这些结果验证了我们的蛋白质组学分析,表明ATAD3A寡聚化的减少提高了AD小鼠的CYP46A1水平。由于神经元胆固醇转换受损,CYP46A1 缺乏会导致神经元中的胆固醇积累。ELISA分析显示,5XFAD小鼠皮质中总胆固醇含量增加,然而,这种效应在杂合 ATAD3A 敲除5XFAD小鼠中显著降低(图 5f)。此外,与用对照肽处理的AD小鼠相比,DA1降低了5XFADhomo小鼠皮质中的胆固醇含量(图 5g)。Filipin是一种常用的荧光探针,用于监测细胞和脑组织中的胆固醇沉积。我们在9个月大的5XFADhomo小鼠大脑中观察到菲律宾结合胆固醇的显著积累,以及相对于WT对应的稳定的表达 Neuro2a细胞。此外,DA1对ATAD3A寡聚化的抑制减少了胆固醇沉积(图 5h,补充图 7c)。
脑胆固醇转化为24(S)-羟基胆固醇 (24-OHC) 代表了大脑中主要的胆固醇消除机制。24-OHC 含量已被用作AD中脑胆固醇失调的标志物。与胆固醇不同,24-OHC可以高速穿过血脑屏障。血浆中超过90%的24-OHC是从大脑中加工出来的,这使得血浆24-OHC浓度成为监测大脑胆固醇转换和神经元CYP46A1的有用标志物活动。作者从DA1或对照肽TAT处理的9个月大的WT和 5XFADhomo小鼠收集血浆。5XFADhomo小鼠血浆中24-OHC的浓度降低,反映了脑胆固醇消除的抑制作用。重要的是,持续使用DA1肽处理5XFADhomo小鼠后,24-OHC血浆浓度恢复到与使用对照肽处理WT小鼠相似的水平(图 5i),进一步支持了ATAD3A寡聚在AD小鼠大脑中抑制CYP46A1的作用。同时,我们使用气相色谱-质谱 (GC-MS) 分析了杂合ATAD3A敲除5XFAD和DA1处理的5XFAD小鼠皮质中的脑甾醇。在所有分析的实验组之间,甾醇(例如,羊毛甾醇、酵母甾醇、去甾醇和乳甾醇)的水平是相当的(补充图 7d)。因此,ATAD3A 寡聚化通过改变胆固醇代谢而不是生物合成来影响 AD 模型中的脑胆固醇水平。

图5 ATAD3A寡聚化抑制AD模型中的胆固醇转换
ATAD3A寡聚在转录水平上抑制CYP46A1
接下来,作者确定异常的ATAD3A寡聚化如何影响CYP46A1的表达。ATAD3A-WT-Flag或ATAD3A-N50-Flag的过表达增强了ATAD3A寡聚化,显著降低了Neuro2a细胞中的CYP46A1 mRNA和蛋白质水平(图 5j;补充图 7e),但没有改变线粒体ATPB的蛋白质水平和VDAC或MAM蛋白SigmaR1的水平(补充图 7e)。ATAD3A-WT和ATAD3A-N50没有改变CYP51A1和HMGCS1的 mRNA水平,它们分别是参与胆固醇代谢和生物合成的基因(补充图 7f)。在暴露于寡聚Aβ1-42肽或稳定的APP表达Neuro2a细胞的HT-22细胞中,DA1对ATAD3A寡聚化的抑制显著增强了CYP46A1 mRNA水平(图5k)。相反,在相同条件下,不同实验组之间的CYP51A1 mRNA水平相当(补充图7g)。ATAD3A蛋白水平和寡聚化不受CYP46A1过表达或伏立康唑抑制 CYP46A1 酶活性的影响(补充图 7h,i)。因此,ATAD3A作用于CYP46A1的上游,ATAD3A 寡聚化在转录水平上选择性地抑制 CYP46A1。
与表达对照载体的细胞相比,在Neuro2a细胞中过度表达ATAD3A-WT-Flag和ATAD3A-ΔN50-Flag变体增加了总胆固醇含量,降低了24-OHC含量,在表达ATAD3A-ΔN50Flag的细胞中观察到的程度更差(图5l,m,补充图7j)。相反,DA1处理消除了ATAD3A-WT或ΔN50诱导的胆固醇积累,并且ATAD3A-ΔCCFlag变体的过度表达(阻止ATAD3A寡聚体的形成)对胆固醇含量的影响与对照载体表达细胞中的效果相当(补充图7j)。这些数据支持ATAD3A寡聚对胆固醇积累的直接影响。此外,CYP46A1过表达降低了ATAD3A WT Flag和-ΔN50 Flag表达细胞系中胆固醇含量的增加,并纠正了24-OHC水平(图5l,m)。类似地,用CYP46A1的变构激活剂依法韦伦(EFV)治疗也消除了ATAD3A-WT和ΔN50诱导的胆固醇累积(补充图7k)。因此,ATAD3A寡聚诱导的胆固醇稳态扰动依赖于CYP46A1。在涉及胆固醇代谢途径的16个基因中,5XFAD小鼠的LDLR和ApoE mRNA水平显著升高,但在5XFAD小鼠和5XFAD小鼠中均减弱。24-OHC是核肝X受体(LXR)的内源性激动剂,其随后调节LXR靶向基因表达(例如LDLR和ApoE),平衡脑胆固醇稳态。因此,通过ATAD3A杂合敲除或DA1治疗降低AD小鼠的LDLR和ApoE mRNA水平与增强的CYP46A1水平一致。总之,这些数据支持作者的假设,即CYP46A1的缺失至少部分地导致了AD模型中ATAD3A寡聚诱导的神经元胆固醇积聚。
ATAD3A寡聚化以依赖CYP46A1的方式促进APP加工
体内对CYP46A1缺陷的补偿可减少AD小鼠的淀粉样沉积并改善空间记忆。CYP46A1过度表达提供的神经保护机制被认为可降低膜脂筏中的胆固醇含量,并减少淀粉样变性APP处理。在AD患者死后皮层中,作者观察到增大和肿胀的脂筏,其抗霍乱毒素B五聚体(CTxB)免疫阳性,CTxB是一种脂筏标记物。此外,CTxB阳性脂筏与APP共定位(补充图8a),这与APP在脂筏中富集以进行处理的概念一致。在5XFAD小鼠大脑中,CTxB阳性脂筏的强度在皮质第五层增加,并与APP蛋白表达增加共定位(补充图8b)。
5XFADhet小鼠中的杂合ATAD3A敲除或DA1介导的ATAD3A寡聚化抑制减弱了CTxB免疫密度(图 6a,b),表明脂筏正常化。因为AD患者和AD 5XFAD 小鼠大脑中APP免疫阳性细胞的ATAD3A强度增加(图 1j),我们检查了APP的加工是否受到异常ATAD3A寡聚化的影响。由ATAD3A-WT-Flag或ATAD3A-N50-Flag过表达介导的增强的ATAD3A寡聚化加剧了C99片段的产生,C99片段是一种存在于稳定的表达APP[wt]的Neuro2a细胞中的APP的病理性蛋白水解产物。相反,ATAD3A 寡聚化被任一ATAD3A-CC-Flag变体的表达或用DA1处理消除了C99的产生(图 6c)。DA1处理还消除了稳定的表达APPwt和 APPswe的 Neuro2a 细胞中的 C99 片段(补充图 8c)。此外,我们观察到 5XFAD 小鼠大脑中 C99 片段水平显著增加,杂合 ATAD3A 敲除或 DA1 处理则降低了这一水平(图 6d)。因此,抑制ATAD3A寡聚能减少AD模型中的APP加工,与减少 5XFADhet;CMV;ATAD3Afl/ 小鼠和DA1处理的5XFADhomo小鼠中的淀粉样蛋白聚集结果一致(图 3,4)。

图6 ATAD3A和CYP46A1合作推动APP加工
作为细胞内脂筏的一部分,MAM为APP处理提供了关键的信号平台。最近的蛋白质组学分析表明,参与胆固醇代谢和Aβ清除的蛋白质(例如CYP46A1和ABCG1)存在于MAM上,并在AD的早期症状前阶段发生改变。尽管CYP46A1在内质网中富集,但在小鼠大脑的MAM部分中可以检测到CYP46A1(补充图8d)。在WT小鼠脑中也观察到CYP46A1和VDAC之间的PLA阳性斑点(补充图8e)。此外,ATAD3A和CYP46A1在WT小鼠的大脑中相互作用,而5XFAD小鼠大脑中的相互作用减少,很可能是由于AD小鼠大脑中CYP46A1的丢失(补充图8f)。这些数据表明,MAMs上的CYP46A1与ATAD3A形成复合物。为了确定CYP46A1介导的ATAD3A寡聚化的缺失是否诱导APP加工,作者在存在DA1肽或控制肽TAT的稳定APP表达神经2A细胞中过度表达CYP46A1或控制载体。与用DA1肽处理的稳定表达APP的Neuro2a细胞观察到的结果类似,在稳定表达APPwt和 APPswe的Neuro2a细胞(图6e)中,仅CYP46A1的过表达可消除C99片段水平,表明APP处理受到抑制。DA1介导的ATAD3A寡聚抑制以及CYP46A1的过度表达对C99片段水平没有加性影响(图6e)。这些结果与作者的观察结果一致,即EFV治疗消除了ATAD3A WT或-ΔN50诱导的稳定APPwt Neuro2a细胞中C99生成的增加(补充图8g)。总的来说,作者的研究结果表明,在 AD 中,ATAD3A 与 CYP46A1 合作介导 APP 处理,可能是在 MAM 中。
ATAD3A寡聚化导致AD模型中的突触丢失
在 AD 病理学中观察到突触丧失,脑胆固醇代谢的破坏已被证明会导致突触丧失和随后的认知缺陷。作者评估了异常的ATAD3A寡聚化是否影响突触是通过使用抗synaptophysin(一种突触前蛋白)和抗PSD95(一种突触后蛋白)共染色来量化突触密度的。在原代小鼠皮质神经元中,Flag标记的ATAD3A-WT或ATAD3A-N50 的过表达降低了synaptophysin和PSD95沿树突的共定位,反映了突触密度的降低和神经元细胞死亡的增加(图 7a,补充图 9a)。此外,用寡聚Aβ1-42肽处理原代神经元降低了突触密度并诱导细胞死亡(图7b,c)。这些效应通过ATAD3A敲低或DA1处理得到纠正(图7b,c,补充图 9b)。此外,单独的CYP46A1过表达增加了暴露于寡聚Aβ1-42肽的原代神经元的突触密度。与单独的DA1处理或CYP46A1过表达相比,阻断ATAD3A与DA1寡聚化然后CYP46A1过表达对沿MAP2 树突的突触素阳性簇的数量没有额外的影响(图 7d)。类似地,通过EFV处理增强CYP46A1消除了ATAD3A-WT-或ATAD3A-N50诱导的原代神经元中的突触损失(补充图9c)。这些结果进一步支持了 CYP46A1介导 ATAD3A寡聚化诱导的神经元的假设。即通过损害胆固醇周转对 AD 造成损害。
最后,作者评估了 ATAD3A 寡聚化对 AD 小鼠突触蛋白和突触形态的影响。蛋白质印迹分析显示 5XFAD小鼠皮质中的突触素和PSD95蛋白水平显著降低;然而,它们的水平可以通过杂合ATAD3A敲除或DA1处理恢复(图 7e,f)。通过高尔基-考克斯染色评估的神经元脊柱密度在9个月大的5XFADhomo小鼠中降低。值得注意的是,与用对照肽处理的5XFAD小鼠相比,在年龄匹配的AD小鼠中用DA1治疗增加了树突棘的数量(图 7g)。因此,抑制ATAD3A寡聚化可减少5XFAD小鼠的突触损失,表明通过遗传或药理学抑制ATAD3A寡聚化可改善AD小鼠的认知活动(图 3、4)。

图7 ATAD3A 寡聚化导致 AD 模型中的突触丢失
小 结
在这项研究中,作者证明了ATAD3A的病理性寡聚化上调了ER-线粒体连接,损害了胆固醇稳态,并促进了淀粉样蛋白的加工,导致AD中的神经退行性变。此外,作者发现了介导AD病理学和认知缺陷发展的ATAD3A-CYP46A1-APP 信号轴(补充图 9d)。因此,我们的研究结果为AD的发病机制提供了见解,并证明ATAD3A 寡聚化是治疗AD和其他与MAM超连接性和胆固醇紊乱相关的神经系统疾病的潜在治疗靶点。

补充图 9d 介导AD病理学和认知缺陷发展的ATAD3A-CYP46A1-APP 信号轴
精读文献请阅读原文:
Yuanyuan Zhao, Di Hu, Rihua Wang, Xiaoyan Sun, Philip Ropelewski, Zita Hubler, Kathleen Lundberg, Quanqiu Wang, Drew J Adams, Rong Xu, Xin Qi. ATAD3A oligomerization promotes neuropathology and cognitive deficits in Alzheimer's disease models.Nat Commun. 2022; 13(1):1121.
.
编 译 / 陈瑞年
校 审 / 蔡玉洁

免责声明:本文仅代表文章作者的个人观点,与本站无关。其原创性、真实性以及文中陈述文字和内容未经本站证实,对本文以及其中全部或者部分内容文字的真实性、完整性和原创性本站不作任何保证或承诺,请读者仅作参考,并自行核实相关内容。文章投诉邮箱:anhduc.ph@yahoo.com






