钯碳还原双键:两次C-H键活化实现钯催化的
环加成反应作为有机合成的一种重要工具,广泛应用于天然产物全合成、药物化学、农药化学等领域,其中最经典的要数[4 2] Diels-Alder反应。因为基于轨道对称理论的环加成涉及双键,起始原料多为偶数烯烃。但是对于含奇数碳原子的环戊烷合成所需的[3 2]环加成反应,设计合适的三碳单元并实现环加成反应却颇具挑战(图1A)。事实上,已有一些报道可以直接生成游离的三亚甲基甲烷(TMM)中间体(即[3 2]反应的三碳单元),但是该反应只针对特定的底物,适用性十分有限。在此基础上,化学家们开发了用过渡金属催化剂产生TMM-等价物的策略,如Trost等人率先将Pd-TMM配合物用作各种[3 2]环加成反应的中间体,并将其扩展到环戊烷的不对称催化中。然而,制备这些TMM-等价物仍需要通过环张力或多次预官能团化来高度活化前体。尽管目前还开发了其它的环加成方法来合成环戊烷,但需要在底物上安装多个π键或离去基团。
1997年,Catellani报道了降冰片烯接力参与的1,2-C(sp2)-H键活化(Angew. Chem. Int. Ed., 1997, 36, 119),也就是现在广为熟知的Catellani反应。受此启发,美国斯克里普斯研究所(The Scripps Research Institute,TSRI)余金权教授课题组设想能否将类似的接力C(sp3)-H键活化用于简单易得且未官能团化的三碳丙烷骨架来实现[3 2]环加成反应(图1B)?近日,他们通过两次C(sp3)-H键活化实现了钯催化的[3 2]环加成反应。具体而言,在钯催化的作用下,脂肪族酰胺先进行β-C(sp3)-H键活化,然后插入马来酰亚胺,触发了第二次C(sp3)-H键活化,最终实现了[3 2]环加成反应。该反应的关键在于使用弱配位的酰胺作为导向基团,这是因为较强配位的导向基与马来酰亚胺反应时会优先发生Heck反应或烷基化反应。相关成果发表在J. Am. Chem. Soc.上。
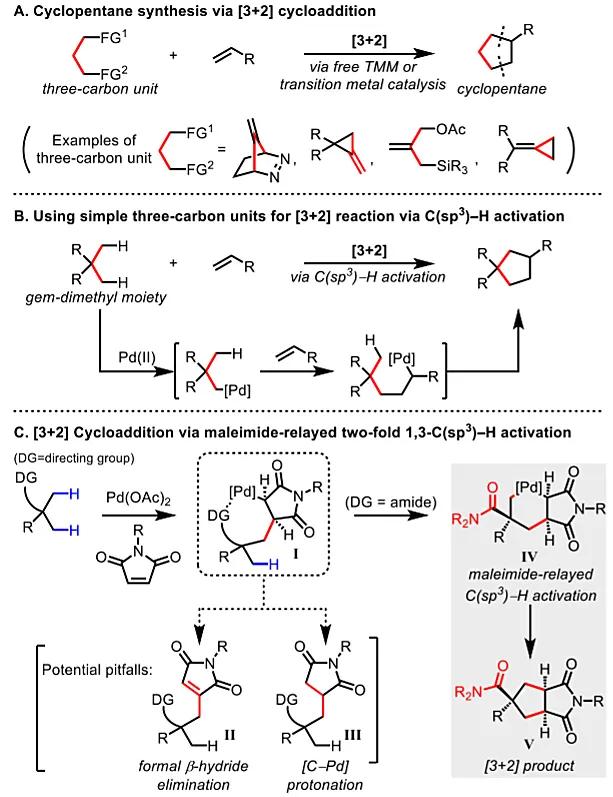
图1. [3 2]环加成反应。图片来源:J. Am. Chem. Soc.
作者选择马来酰亚胺作为中继烯烃而不是降冰片烯,主要有以下两方面原因:(1)与降冰片烯得到的产物相比,马来酰亚胺得到的产物具有显著的合成效用;(2)马来酰亚胺是反应性环状烯烃,已被广泛用于Pd-催化的C(sp3)-H键官能团化的偶联试剂。然而,插入马来酰亚胺后的关键中间体(I)可能会经历不希望的β-H消除(Heck产物II)或质子化(烷基化产物III),而不是接力C(sp3)-H键活化(图1C)。有意思的是,采用新的导向基或配体或许可以调控物种(I)再次进行C(sp3)-H键活化。他们推测使用弱配位的导向基(酰胺)可能会促进C(sp3)-H键的接力活化,因为Pd中心更容易从导向基中释放出来,从而实现第二次C(sp3)-H键活化。
首先,作者选择新戊酰胺1与N-(4-硝基苯基)马来酰亚胺作为模板底物,考察了不同的配体(图2)。当不加配体时,环戊烷产物的收率仅为10%,同时观察到痕量的Heck产物(< 5%)。当使用吡啶3-磺酸(L3)作为配体时,可显著提高产率(65%);当使用更缺电子的L4时,反应的收率高达87%。然而,无论使用哪种配体,都会生成痕量的Heck产物。值得一提的是,当新戊酸与L4在相同的条件下进行反应时,仅观察到Heck产物,这表明使用弱配位的酰胺导向基对于[3 2]环加成反应至关重要。此外,作者还分析了该反应的立体化学。首先,关于稠合的双环核,[3 2]环加成产物显示出cis-环连接,证实了第二次C(sp3)-H键活化以接力的方式发生。其次,主要非对映异构体的构型在酰亚胺基团和导向基之间呈现出cis,而次要非对映异构体则呈现出trans,这表明第二个C(sp3)-H键活化步骤在剩余的两个甲基之间是有选择性的。

图2. 配体的筛选。图片来源:J. Am. Chem. Soc.
在最优条件下,作者考察了[3 2]环加成反应的底物范围(图3)。首先,考察了酰胺上的取代基(图3A)。简单的烷基(2-3)、芳基(4)、杂原子官能团(5-8)以及饱和杂环(9-12)取代的底物都能兼容该反应,以中等至较好的收率(56-80%)和非对映选择性(dr 2:1-9:1)得到所需的环戊烷产物,但带有α-H的底物却不能兼容该反应。接着,他们考察了氨基上取代基的范围(图3B)。不同位阻的二烷基氨基(13–15)以及环状的二烷基氨基(16–21)都成功地得到了相应的产物。氨基酸衍生的底物(22–23)、Weinreb酰胺(24)以及N-芳基取代(25)的底物也能以良好的产率(64-78%)得到所需的产物。虽然仲酰胺(26-28)也能实现这一转化,但必须使用缺电子的马来酰亚胺才能实现高收率。一般而言,与缺电子的马来酰亚胺(活性更高的Michael受体)相比,简单的N-苯基马来酰亚胺的反应活性较低,特别是由于Thorpe-Ingold效应,仲酰胺的收率要比叔酰胺低。
此外,作者还系统地研究了马来酰亚胺N-取代基的底物范围(图3C)。对于N-芳基马来酰亚胺而言,不论对位带有富电子取代基(29-30)还是缺电子取代基(31-35),甚至邻位和间位带有取代基(36-37)的底物均具有较高的收率(63-84%)和非对映选择性(dr 4:1-10:1)。富电子的N-杂芳基马来酰亚胺(38-39)也能兼容该反应,尽管非对映选择性略低。N-烷基马来酰亚胺也具有较高的反应活性,无论N-取代基的电性和位阻如何(40-44)。但是,当测试其他相关的缺电子烯烃(如马来酸酐)时,未观察到[3 2]环加成产物。最后,他们考察了内酰胺的底物范围(图3D)。值得一提的是,六元内酰胺(45)以47%的收率得到所需的双环产物。七元内酰胺甚至更大环的内酰胺(46–48)也能实现[3 2]环加成,以较高的收率(71-79%)得到双环产物。另外,官能化的七元内酰胺(49-51)以及1,4-二氮杂酮衍生的底物(52)也适用于[3 2]环加成反应。总的来说,所有情况下得到的主要非对映异构体,其酰亚胺基团和酰胺导向基之间呈现cis-构型。

图3. 底物扩展。图片来源:J. Am. Chem. Soc.
为了进一步证明该反应在有机合成中的应用,作者进行了克级规模制备,以79%的收率得到产物20(图4A)。当用廉价的铜盐作为氧化剂时,也可以克级规模进行[3 2]环加成反应,尽管收率较低(46%)。此外,根据Jones等人开发的方法,他们用(1S,2R)-cis-1-氨基茚满-2-醇衍生的催化剂对[3 2]环加成产物20-maj进行还原去对称化,得到羟基内酰胺53,后者可以直接转化为内酰胺54(98% ee),尽管收率仅为45%(brsm),这是由于过度还原导致吡咯烷副产物的生成。

图4. 克级规模反应及环戊烷产物的还原/去对称化。图片来源:J. Am. Chem. Soc.
另一种可能的机理途径是先形成Heck产物,然后再进行第二次C(sp3)-H键活化/分子内迁移。为了验证该机理的可行性,作者制备了Heck产物55(图5)。无论是否存在马来酰亚胺,都未观察到[3 2]环加成产物20的形成,这意味着Heck产物不是[3 2]环加成反应途径的中间体。

图5. Heck产物的反应尝试。图片来源:J. Am. Chem. Soc.
总结
余金权教授课题组通过两次C(sp3)-H键活化,开发了含偕二甲基酰胺底物和马来酰亚胺之间的[3 2]环加成反应,该反应的关键在于弱配位的酰胺导向基。为了实现酰胺导向的C(sp3)-H键活化,至关重要的是使用缺电子的吡啶-3-磺酸。此外,作者还在手性噁唑硼烷催化下,实现了[3 2]环加成产物的还原去对称化,以优异的对映选择性得到手性环戊烷。鉴于这两个C(sp3)-C(sp3)键都是从C(sp3)-H键活化直接构建而成,无需预官能团化,因此该方法将是常规环戊烷合成策略的重要补充。
Palladium-Catalyzed [3 2] Cycloaddition via Two-Fold 1,3-C(sp3)-H Activation
Hojoon Park, Jin-Quan Yu
J. Am. Chem. Soc., 2020, DOI: 10.1021/jacs.0c08290
导师介绍
余金权
https://www.x-mol.com/university/faculty/694
,免责声明:本文仅代表文章作者的个人观点,与本站无关。其原创性、真实性以及文中陈述文字和内容未经本站证实,对本文以及其中全部或者部分内容文字的真实性、完整性和原创性本站不作任何保证或承诺,请读者仅作参考,并自行核实相关内容。文章投诉邮箱:anhduc.ph@yahoo.com






