硫代乙酰胺的合成路线图(通过三磷酸盐介导的α-胺化的羰基1)
通过三磷酸盐介导的α-胺化的羰基1,2-转位反应
文章出处:Zhao Wu, Xiaolong Xu, Jianchun Wang, Guangbin Dong. Carbonyl 1,2-transposition through triflate-mediated α-amination. Science 2021, 374, 734-740.
摘要:到目前为止,在给定的分子骨架内选择性迁移羰基氧仍然是一个挑战,特别是邻近的碳。在这项工作中,作者描述了一个简单的一或二锅方案,将酮转位到邻近的碳。该方法首先将酮转化为相应的三氟烯酸烯基,然后在双功能氢和氮供体的催化下,进行Pd和降冰片烯催化的区域选择性α-胺化和异丙醇ipso-加氢。由此产生的“转座烯胺”中间体随后可被水解,产生1,2-羰基迁移产物。这种方法允许通过后期功能化快速获得不寻常的生物活性类似物。
羰基在化合物中的特定位置可以强烈地影响化合物的生物学和物理性质,以及其作为合成中间体的战略用途。例如,熊果酸中C3-OH转位到相邻的C2位置(图1A)导致抑制糖原磷酸化酶的效能提高13倍。同样地,由降钙蒎酮的C2-羰基异构体衍生的化合物对氨基糖苷诱导的听力损失的活性比相应的C3类似物增加了9倍。从合成规划的观点来看,高效的羰基1,2-置换方法也可以简化复杂目标分子的构建,允许与更容易接近的底物策略性的键断开;例如,卡藜酮和Lycoraminone采用更有效的羰基1,2迁移策略,总合成将被简化(图1B)。
羰基1,2的转位不是一个简单的过程(图1C)。目前,最普遍的策略是使用酮的α-功能化来引入羰基替代品,然后是一系列的下游转换。可选择的策略包括形成一个三元环或1,2-二酮中间体,这些中间体可以不对称地产生正式的羰基迁移产物。除了需要长合成序列外,底物特异性和区域选择性是与不对称方法相关的额外问题。因此,具有广泛功能基团(FG)耐受性的一般、区域选择性和直接的羰基1,2转位方法仍然是一个重要的目标。鉴于从羰基化合物中容易形成烯基磺酸盐(如三氟酸盐),作者设想了开发三氟酸盐介导的α-胺化过程,以获得转置的烯胺中间体,该中间体可以进行原位水解,生成羰基1,2迁移产物(图1D)。关键的烯胺形成步骤包括同时加成氢化物到配位(三氟酸盐所在位置)和氮取代基到邻近的烯基碳(α位置)。作者的目标是通过Pd和降冰片烯(Pd/NBE)的协同催化,实现三氟烯基的α-胺化和异丙醇ipso-加氢。

图1
Pd/NBE协同催化最初由Catellani发现,自1997年以来广泛应用于芳烃功能化。相比之下,它在非芳香族底物功能化中的应用非常罕见;特别是,它还没有被用来在烯基底物上引入杂原子取代基。为了通过Pd/NBE催化实现三氟烯烃的α-胺化和异丙醇ipso-加氢,必须解决两个主要挑战(图2)。首先,在与Pd(0)氧化添加和最终生成的烯基-Pd(II)迁移插入NBE后,以避免产生副产物B(已知的主要竞争途径)的3-外三角环化,NBE共催化剂必须有一个刚性取代基,如C2位置的酰胺基团(与Pd结合的碳),以强烈促进关键烯基降冰片基Pd环(ANP)的形成。虽然NBE上增加的空间位阻可能不会阻碍与体积较小的亲电试剂(如伯烷基卤化物)的反应,但当使用空间要求更高的胺亲电试剂时,它可能会大大阻碍反应。缓慢的胺化会导致过早的氢化物终止(形成副产物A和C),以超过所期望的途径。第二,即使C-H胺化确实发生,所产生的烯胺部分也会通过氮孤对电子或富电子π键与Pd(II)强烈配合,这与碳基亲电试剂的类似反应相反。因此,NBE (β-碳消除步骤)的挤压会更加困难,这可能会产生副产物D。因此,明智地选择NBE助催化剂和仔细设计胺试剂可能是平衡每个基本步骤的要求和解决上述挑战的关键。
作者假设在NBE上有适度的空间位阻有利于抑制环丙化途径,促进β碳的消除,而不抑制烯基-降冰片基Pd环与亲电试剂之间的反应(避免产生副产物A和B)。此外,需要一个体积大的氢化物源,特别是可以缓慢释放氢化物的源,以尽量减少早期氢化物终止(避免产生副产物C和D)。为了验证这一假设,研究人员调查了一系列NBE助催化剂,并设计了含有亲电胺基团和掩体氢化物的双官能碳酸酯基试剂。当以α-四酮(化合物1)为模型底物时,经过对各种反应参数的仔细评估,最终在一锅法中获得了所需的β-四酮(化合物3),产率为88%。通过反应物的连续加成,三氟烯酸烯基中间体(化合物2)原位形成,并进行随后的转位胺化和水解,而不需要分离或改变反应容器。关键是确定有效的三氟化的反应条件,并与Pd/NBE催化兼容。以三氟酸酐为三氟化试剂,在甲苯和1,4-二恶烷的混合溶剂中以Cs2CO3为碱是最优的,可获得接近1倍当量的三氟化烯(化合物2)。单独甲苯的转化率较低。尽管体积庞大的吡啶碱对三氟甲烷的形成是有效的,但它对随后的胺化步骤是有害的,可能是相容性问题的结果。在Pd/NBE催化步骤中,以四甲基哌啶Li (LiTMP)为基料形成反应平稳的非氟化Li中间体,反应干净,总效率高。这提供了另一种一锅法的条件。
在最佳的三氟化条件下,研究了不同的亲电胺源对α-胺化和异丙醇ipso-加氢反应的影响。常见的亲电性吗啡啉(化合物R1),加上外源性的氢化物源,只能产生24%的化合物3,同时形成大量直接反相还原(A-型)副产物。通过与大体积的醇片段连接,容易接近的吗啡啉衍生碳酸酯化合物R2到R6达到了产率提升很大和抑制副反应的效果,从2,4-二甲基-3-戊醇(化合物R4)衍生的试剂显示出优越的结果。从其它胺(化合物R7到R9)衍生的碳酸盐试剂也进行了测试;在这种情况下,以哌啶为基础的一种化合物R8被发现是最优的,因为相比于其它胺,它可以实现完全转换而副产物可以忽略。对照实验表明,Pd和NBE对该反应都是必要的,而且在没有吡啶酮添加剂的情况下产率下降。在研究的所有结构修饰的NBE中,氮叠氮丁酰胺NBE (化合物N10)被证明是最有效的。当降低N10负载时,观察到类似的产率,显示出一定的催化活性。简单的NBE(化合物N1)和C1-取代的化合物N4生成大量环丙烷(化合物B)和烯胺D-类副产物,与作者之前的观测结果一致。具有C5和C6二取代基(化合物N2和N3)和C2酯基(化合物N5)的NBEs在这一转化中表现较差。N-甲基(N-Me)酰胺NBE(化合物N7)在形成NBE的副产物时只能获得中等的产率,而体积较大的二甲基和二乙基酰胺NBE则导致反应活性大幅下降,形成更多的副产物A,并发生均偶联反应。化合物N10的高反应活性和选择性在更具有挑战性的底物中表现得更加明显,如非共轭三氟烯基(化合物18a)。此外,Buchwald的Ph-DavePhos配体(Ph = 苯基)效率最高。虽然富电子的Ph-JohnPhos (配体L2)略少,但产率略低,常规davepos (配体L3)和PPh3 (配体L4)的反应活性大大降低。
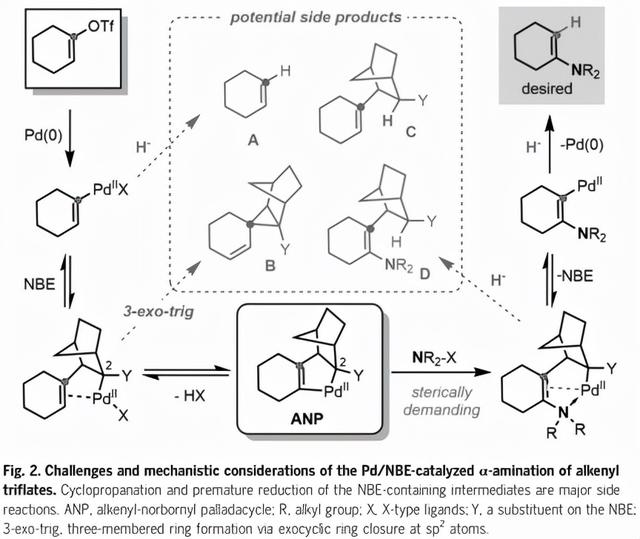
图2
在优化的条件下,作者进一步考察了羰基1,2转位反应底物的范围(图3)。一系列在脂肪环(化合物5和6)和芳香环(化合物8-12和14)上都有取代基的α-四酮衍生物可以通过一锅法反应得到所需的β-四酮。色罗马酮(化合物6)也发生反应,尽管产率较低,可能是由于杂原子的配位能力。除了四酮外,由五元1-茚酮(化合物15)、七元1-苯并环庚酮(化合物16)和取代苯并环庚酮(化合物17)衍生的底物也提供了所需的解共轭产物。当相应的三氟烯基中间体对空气敏感时,如前驱体对化合物8。对于硫色酮(化合物7)和含有高度缺电子的硝基芳烃(化合物13)的底物,当三氟烯基中间体被提纯时,总产率要高得多。此外,非共轭酮类也被证明是合适的底物。虽然一锅法(以LiTMP为基础)以一个有用的产率提供了所需的羰基1,2移位产物(如化合物18),但两步法通常导致更高的整体效率。在γ位上有取代基的环己酮被转化成相应的β-取代环己酮(化合物18-21),与烯酮共轭加成反应互补。相反,β-取代环己酮能以较高的产率转化为γ-取代环己酮(化合物22-26),这对于通过传统方法制备环己酮来说是很重要的。尽管烯酰化步骤的区域选择性从4.7: 1到11: 1不等(有利于在空间阻碍较小的一侧去质子化),但胺化几乎完全发生,在所有情况下(化合物22-26和37)产生主要的(较小的)异构体。值得注意的是,端烯烃(化合物22)保持完整,在这些条件下没有观察到异构化。具有α-季碳中心(化合物27,30和31)、α,β-环丙烷(化合物28)和α-酯(化合物29)取代基的环己酮也反应生成所需的产物,这些产物很难通过其它方式得到。此外,由Pauson-Khand反应衍生的α-取代环戊酮(化合物32)和5-5双环酮(化合物33)被证明是有效的底物。各种FGs,包括芳基氯(化合物11)、腈(化合物10)、硝基(化合物13)、苄醚(化合物20)、磺酰胺(化合物21)、烯烃(化合物22,30和34),缩醛(化合物31)、酯(化合物12-28)、内酯(化合物34和38)、噻吩(化合物8和14)、喹唑啉(化合物23,24)、茶碱(化合物25)和吲哚(化合物26),都符合这些条件。此外,从天然产物如托品酮(化合物35)、诺品酮(化合物36)、猪去氧胆酸(化合物37)和α-三托宁(化合物38)中得到的三烯基三托酯通过平滑反应得到相应的羰基迁移类似物。

图3
如前所述,由于有利的烯醇化过程,β-取代环己酮转化为γ-取代类似物很简单;然而,羰基向另一个方向迁移并形成α取代的产物将会更加困难,因为相应的动力学不利烯醇酯通常产率低,选择性差(图4A)。此外,在所需的三氟烯基中存在β取代基会阻碍C-H的Pd化步骤,类似于在芳香底物中观察到的“间位约束”。因此,实现这种“β到α”的转变是困难的。在这一挑战的刺激下,作者构思了基于替代酮的策略(图4B)。将烷基亲核试剂与环己酮底物共轭加成,然后用Comins试剂猝灭,以中等到良好的产率生成所需的三氟烯基。其次,考虑到β位位阻,作者假设较小体积的胺亲电试剂应该有利于C-H胺化步骤。使用氮叠氮丁衍生的亲电试剂(化合物R10,预计比二甲胺还要小)是最理想的。β到α的迁移产物(化合物40和41)完全保留了相对立体化学结构。这些α, β-二烷基化产物如化合物40和41以前没有报道过,可以作为有用的合成中间体。此外,两步法将( )-二甲基双环庚烯酮转化为(-)-松莰酮(化合物42)。此外,该策略对于制备对映体富集的α-烷基化酮尤其有用,因为直接不对称非烯丙基α-烷基化酮仍然具有挑战性。例如,通过利用完善的对映选择性共轭烷基加成(化合物36),α-乙基酮(化合物43)通过酮1,2-迁移策略获得了优异的对映选择性(图4C)。

图4
最后,探索了该方法的综合应用(图5)。与富电子二烯的Diels-Alder反应是环己酮合成方法中应用最广泛的方法之一,并已在众多的全合成中得到应用。例如Danishefsky的二烯与Michael受体反应,得到C4位置吸电子基团的环己酮。在这项工作中,通过原位捕获硅基烯醇醚中间体作为相应的三氟烯酸烯基,高效率地获得了具有相反区域选择性的Diels-Alder产物(位于C3位置的吸电子基团) (图5A)。该序列提供了一种独特的、模块化的方法来获取多取代环己酮,而Diels-Alder策略是无法实现的。除了质子,其它亲电试剂也可用来捕获烯胺中间体。例如,烷基化酮1,2-迁移是通过使用溴化烯丙基作为亲电试剂生成ipso C-C成键产物(化合物46) (图5B)。另外,不对称共轭加成后的羰基1,2-迁移可以通过对映选择性的方式引入γ-立体中心。这种方法在食欲素受体拮抗剂的不对称合成中得到了例证(化合物50),这是之前通过手性拆分方法制备的(图5C)。在这种方法中,醇中间体(化合物49)可以通过已知的非对映选择性还原富对映酮(化合物48),该富对映酮(化合物48)是通过不对称共轭加成和酮1,2-迁移序列合成的,具有良好的总产率。
此前,二氢睾酮醋酸酯和二氢胆固醇甾体经五步转化为具有生物活性的“C2-oxo”类似物,总产率分别为< 39%和10%(图5D)。前者的区域选择性较差(1: 2.2),使产物的纯化复杂化。使用相同的原料,这种羰基1,2转位法允许两步合成这些产物为单一区域异构体,产量较高(化合物51和53)。类似地,“C2-oxo”齐墩果酸衍生物,它是一种糖原磷酸化酶抑制剂,之前是通过三步序列制备的,总产率为37%,但选择性差。相比之下,作者的方法可以用更少的步骤,更高的总产率和完全的区域选择性提供相同的产品(化合物52)。最后,Pallescensin A全合成的关键中间体反式-萘烷酮(化合物54)先前由Wieland-Michele酮方法分8步合成,总产率21% (图5E)。通过比较,作者从现成的香叶酰溴开始,只用三步就合成了相同的酮中间体(化合物54),总产率为32%。鉴于这种酮转位方法的便利性和广泛的FG耐受性,作者预期它将在复杂分子的合成和FG转位类似物的高效制备中得到广泛的应用,这将有利于药物化学研究。

图5
,免责声明:本文仅代表文章作者的个人观点,与本站无关。其原创性、真实性以及文中陈述文字和内容未经本站证实,对本文以及其中全部或者部分内容文字的真实性、完整性和原创性本站不作任何保证或承诺,请读者仅作参考,并自行核实相关内容。文章投诉邮箱:anhduc.ph@yahoo.com






