锂离子电池层状结构正极材料分类(层状结构正极材料的发展历程之病秧子层状锰酸锂)

如果能全部采用锰作为锂电正极材料,那资源不是问题、价格不是问题、甚至环境也不是问题(锰毒性远比钴镍低得多),但锰有自己 的问题—无论是层锰还是尖晶石锰酸锂都是病秧子、前一个结构不稳,难以合成;后一个在高温循环中性能迅速衰减。
元素可以形成LiMnO2和LiMn2O4两种结构电活性正极材料,前种结构类似LCO,属于层状结构,简称层锰或以符号LMO-layer表示,电压平台3.6V,理论比容量285mAh/g;后者则是业界非常熟悉并已经大量应用的尖晶石锰酸锂,可用符号LMO-spinel表示,电压平台4V左右,理论比容量148mAh/g,本章节主要讨论层状锰酸锂LMO-layer。
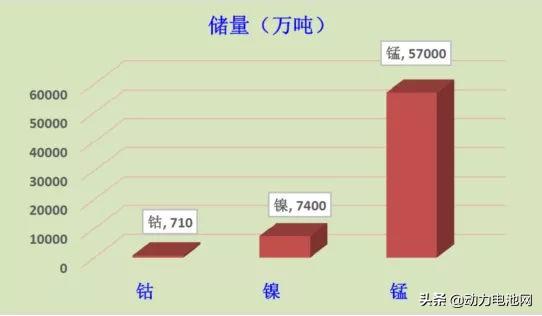
在目前已探明的资源储量中,钴的储量约为710万吨(USGS,2016),镍的储量为7400万吨(USGS,2015),而锰的储量为5.7亿吨(USGS,2015),如果再考虑到海底的锰结核,则锰资源量可能超过了150亿吨——虽然钴镍锰三兄弟在元素周期表上的位置很近,但家底却以数量级形式递增。
所以,如果能全部采用锰作为锂电正极材料,那资源不是问题、价格不是问题、甚至环境也不是问题(锰毒性远比钴镍低得多),但锰有自己的问题—无论是层锰还是尖晶石锰酸锂都是病秧子、前一个结构不稳,难以合成;后一个在高温循环中性能迅速衰减。用流行的一句话反过来说——上帝给你开了扇窗,却同时把门给你关上了!
与镍的情况正好相反,锰一般以 4价最为稳定,所以LiMnO2中的 3价Mn3 也非常易于在锂离子脱嵌过程中形成 4价Mn4 ,哪怕是锂离子全部脱嵌出去形成仅有锂空位的“□MnO2”(锰的化合价全变成 4价),从化合价上来看也是稳定,毕竟自然界软锰矿(主要成分MnO2)多的是,而对比之下,自然界找不到含CoO2或NiO2的矿物。
层状锰酸锂的主要问题是合成困难以及在循环中的结构不稳定。金属锰原子的外围电子排布是3d54s2,从由氧形成的八面体配位场中,Mn4 八面体场稳定性高于Mn3 八面体场,前者具有更好的对称性。
如下图所示,失去4个电子变成Mn4 后外围就剩下3d轨道的3个电子,在此情况下3个d电子恰好填入三个低能级t2g轨道(dxy、dxz、dyz),保持了轨道的完美对称性。但如果是失去3个电子变成Mn3 后外围3d轨道剩下的是4个电子,受电子成对能限制,4个电子仍然以高自旋方式(单电子)分布在4个d轨道上,这就使得原先处于高能级

的轨道进一步与低能级的d轨道进行重新的能级分裂……

简单说一下,在Mn3 形式下的4电子时代,除了原先的3个d电子尚能保持一定的对称分布,第4个d电子更多地向z轴方向集中分布,使得原本标准的锰氧正八面体被拉长14%左右,而形成了一种扭曲的菱方结构。
具体表现在,晶体结构上的结果,就是主晶层与间晶层从原本的规则层状交叉排列转变为以波纹状交叉排列,是典型的热力学不稳定结构。因此,层锰LiMnO2在现实中的合成难度可想而知。

为此,层状锰酸锂的合成一般是用离子半径更大的钠先合成层状α-NaMnO2,然后用离子交换的办法,让钠离子下来,锂离子上去得到层状LiMnO2。

固相法合成层锰一般是直接使用 3价的锰原料(例如γ-MnOOH),也可以使用 4价的锰原料,然后在反应中还原到 3价。但需要注意的是 3价的锰对氧气非常敏感,极易接触氧气形成其它的锂锰氧化物,需要全程高真空(0.1Pa)和高温(900~1050℃),同时合成完毕后需要快速冷却到Mn3 的氧化活性温度以下,避免 3价锰的氧化,否则会发生以下副反应:

不但如此,即使合成了层锰,其结构在充放电循环中也容易逐渐发生层状到尖晶石型的转变,也是经不起革命长期考验的病秧子。
层锰的优势不仅仅在于资源优势,因为 4价的锰为稳定价态,所以LiMnO2可以在较低充电截止电压下表现出更大的锂脱嵌深度。1996年Armstrong(中文直译:胳膊有劲儿,意思是能拧过大腿?)首次报道了首次放电容量高达270mAh/g的LiMnO2的合成与性质,相当于95%的锂脱嵌深度。
“较低的充电截止电压下具有更大的锂脱嵌深度”—这句话也意味着LiMnO2在平均电压方面的降低。实际上,在LiMO2系列的材料中,其平均工作电压是从钴、镍、锰依次降低的,LCO的平均工作电压为3.8V,LNO平均工作电压只有3.7V,而层锰LiMnO2在同样的比容量下的平均工作电压只有3.6V。
众所周知,材料的比能量是电压与比容量的乘积,所以通过简单数学关系不难推断,与其开发一款比容量200mAh/g的LiMnO2材料,还不如开发比容量为180mAh/g的高压LCO。因为,只有开发具有更高比容量的层锰材料才能凸显其在比能量方面的更大优势。
提到材料电压与比容量关系,我想起一个笑话,某年曾经有主管部门发布项目指南,鼓励发展高比能量正极材料,其中要求材料比容量大于等于180mAh/g,而没提电压的要求。这就好比我们在论证一个水电站项目时只考虑水蕴藏量而不考虑落差一样,结果只能得出在海边修建水电站是最佳方案。
要知道,一般的橄榄石材料理论的比容量才170mAh/g,而尖晶石材料理论的比容量才148左右,这不是象“萝卜招聘”一样的“萝卜项目指南”吗?如果纯粹为了打项目主管部门的脸的话,其实完全可以用硅碳、锡这类材料去申报,你不就是要容量大吗?我4200mAh/g的比容量够不够?因此,针对LiMnO2的高比容量也要结合其平均电压平台来综合考评。
回到本文主题,LiMnO2在使用中也存在着不稳定性——如果将LiMnO2的化学式系数加倍即Li2Mn2O4,这相当于一个锂化尖晶石LiMn2O4结构,特别是对于锂脱嵌50%左右的LiMnO2,基本上与尖晶石LiMn2O4而非常接近了,而后者是一个热力学稳定结构……
所以LiMnO2在使用中会存在一个逐渐向尖晶石相结构转变的不可逆过程。而从下图可以看出,当右图LiMnO2中16c位的锂离子脱嵌出去以后,与左图LiMn2O4的尖晶石结构就非常接近了。

另一方面,锰的稳定价态为 4价,但LiMnO2中的锰其初始平均价态为 3,而Mn3 又是一个非常容易发生歧化反应的离子,进而导致结构失效:

针对LiMnO2的有效改进也是以体相掺杂为最主要的工作,文献报道过的针对层锰的掺杂元素基本上是将常见的元素一一耕耘了一遍,其中有意义的还是与Co、Ni的自家兄弟之间的“互帮互助”,如LiMn1-xNixO2、LiMn1-xCoxO2系列复合氧化物,当然还有LiNi1-xCoxO2系列,统称为二元正极材料。
2001年,Ohzuku首次制备成LiNi0.5Mn0.5O2二元正极材料,在2.5~4.5V之间约有200mAh/g的比容量以及良好的循环性能、安全性能,但材料中大约有8%~10%的Li /Ni2 混排存在,这极大地影响了锂的扩散路径,恶化了材料的倍率性能。当前市场上除了NCA勉强算作二元材料外,还没有其它的二元材料实现商品化应用。
LiMnO2还有一种特殊的掺杂是使用过量的锂替代锰进行的掺杂,即Li(Li1/3Mn2/3)O2,如果消除分母即得到Li2MnO3,其中的锰全部为 4价的Mn4 。从电极材料结构来说,这种掺杂是非常失败的——根据等大球体的六方紧密堆积原理,在1mol的Li2MnO3中,共有3mol的氧八面体空隙和6mol的氧四面体空隙。
显然,这3mol的氧八面体空隙被2mol锂和1mol锰全部占据,已经没有空余的氧八面体空隙接受锂离子的嵌入。另外,从组成元素的化合价态分析,因为锰已经全部是 4价,Li2MnO3也不能进行锂离子的脱嵌(除非锰继续氧化为 5价)。所以,Li2MnO3在2.0~4.5V范围内没有任何电化学活性。
网络上曾听说过一个段子,说孙悟空他爹最厉害,竟然能让石头怀上孕,其实科学家有时候比孙悟空他爹还厉害—有学者经过适当的酸浸、热处理等一系列“回魂大法”后,Li2MnO3竟然在4.8V有了电活性——可以以Li2O的形式整体脱出,剩余氧原子层从原本轻微扭变的四方密堆分布转变为三棱锥与八面体交替分布。
虽然这种电活性的可逆性很差(释氧),但与其它层状材料(LiMO2)复合后,循环性能得到了明显的改善,且具有320mAh/g以上的比容量,这简直是逆天了!要知道,一般的层状材料的理论比容量才280mAh/g左右,—这就是一度神秘兮兮的富锂锰,后面我们会单独进行介绍。
更多精彩内容,敬请点击文末蓝字“了解更多”。
,免责声明:本文仅代表文章作者的个人观点,与本站无关。其原创性、真实性以及文中陈述文字和内容未经本站证实,对本文以及其中全部或者部分内容文字的真实性、完整性和原创性本站不作任何保证或承诺,请读者仅作参考,并自行核实相关内容。文章投诉邮箱:anhduc.ph@yahoo.com






