多肽固相合成法(林旭锋课题组Angew.)
近日, 浙江大学丁寒锋、林旭锋课题组 完成了( )- S arcophytin ( 1 )、( )- C hatancin ( 3 )、(-)-3- O xo c hatancin ( 4 )和(-)- P avidolide B ( 5 )的全合成, 并对(-)- I sosarcophytin ( 2 )进行了结构修正,该成果近期发表在 Angew. Chem. Int. Ed. 上( DOI: 10.1002/anie.201900782 )。

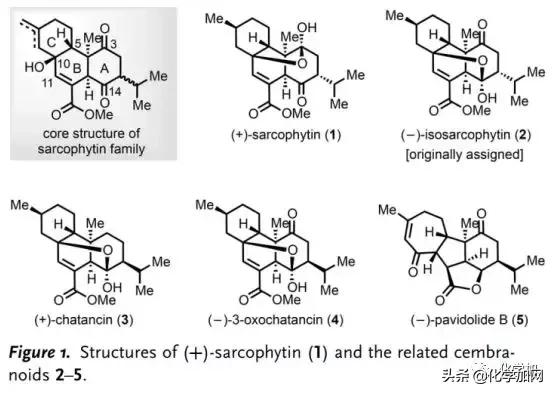
海洋珊瑚中蕴藏着结构多样的西松烷型二萜类天然产物。自从Anjaneyulu课题组于1998年从Sarcophyton elegans中首次分离得到( )-Sarcophytin (1)(Figure 1)以来,至今已分离得到多个Sarcophytin家族成员。西松烷内酯的结构特征在于含有两个具有六个或七个手性中心的顺式十氢萘片段,并通过在C10位叔醇和C3/C14位酮基之间的跨环桥连半缩酮折叠成拥挤的多环骨架;此外,该类天然产物具有潜在的生物活性,如( )-Chatancin (3)是有效的血小板活化因子(PAF)拮抗剂,激发了Gössinger、Deslongchamps、Maimone和Carreira等人对其进行全合成研究。
(图片来源:Angew. Chem. Int. Ed.)
1和3以及(-)-Pavidolide B (5)是由林文瀚课题组从Sinularia pavida中分离得到并鉴定的,其中,5对多种人早幼粒细胞白血病细胞系具有高选择性抑制活性。圆顶状[6,5,5,7]四环体系,特别是全取代的环戊烷母核以及七个连续的手性中心,使其全合成充满挑战。2017年,杨震、龚建贤团队以乙烯基环丙烷的串联自由基环化为关键步骤完成了5的不对称全合成。尽管已经提出了基于分子内Michael/aldol反应的5的生源途径,但通过西松烷跨环Diels-Alder环加成构建的Sarcophytin/Chatancin骨架中B/C环上的C5(10→11)Wagner-Meerwein重排可能是涉及5的另一种生源途径。因此,有必要对这些西松烷内酯的集群式合成进行系统研究,以阐明其多环骨架之间的生源关系。近日,浙江大学丁寒锋、林旭锋课题组完成了( )-Sarcophytin (1)、( )-Chatancin (3)、(-)-3-Oxochatancin (4)和(-)-Pavidolide B (5)的全合成, 并对(-)-Isosarcophytin (2)进行了结构修正。
1-5的逆合成分析如下(Scheme 1):作者认为Sarcophytin家族成员1-4可由反式二醇6或7通过后期官能团化(包括不饱和甲酯的构建、C14位的氧化态调整和/或C1位的立体化学以及半缩醛化)合成。根据5的生源假说,顺式二醇8的频哪醇重排是构建这类天然产物的[6,5,7]三环碳骨架的重要手段。区别主要在C10位手性中心的二醇6-8可以由通用中间体9通过底物控制的Δ9,10-烯烃的面选择性水合来构建,而9可以由双环烯酮10和格氏试剂11通过Helquist环化合成。最后,10可以由易得的手性醇(-)-12和Rawal二烯13通过双重Mukaiyama Michael加成/消除制备。

(图片来源:Angew. Chem. Int. Ed.)
通用中间体9的构建(Scheme 2):作者以(-)-12为原料,经苯甲酰化得到14(95%)后,与Rawal二烯13在40 ℃进行Mukaiyama-Michael加成得到15,为一对异构体(d.r. 2.5:1),通过升高反应温度(70 ℃)显著提高了非对映选择性(>20:1)。将15用HF脱TBS后,进行第二次Mukaiyama-Michael加成,然后消除得到顺式-[6,6]双环二酮10(84%),其C4位季碳的立体化学通过X-射线单晶衍射确证(Scheme 2)。用t-BuOK和碳酸二甲酯处理10经由酯交换/Dieckmann缩合形成中间体16,引入内酯得到17(75%)。烯醇17经“一锅法”Helquist环化[包括铜介导的立体选择性Michael加成(11, CuBr·Me2S, HMPA)和分子内aldol反应(aq. HCl)]得到四环中间体9(82%)。

(图片来源:Angew. Chem. Int. Ed.)
在构建完[6,6,6]三环骨架后,作者将注意力转向Sarcophytin家族成员1-4的全合成(Scheme 3)。利用NaBH(OAc)3对9中烯酮进行化学和非对映选择性1,2-还原得到单一异构体19(76%)。受C11位羟基的位阻影响,Δ9,10-烯烃的水合有利于β-取向选择性。经过广泛的条件筛选,作者采用Shenvi优化的反应条件[Mn(dpm)3, Ph(i-PrO)SiH2, O2, THF]将19水合得到二醇6/8(d.r. 5.6:1,80%)的混合物。反式二醇6经MsCl/DBU处理得到环氧化中间体20,然后经历内酯和环氧开环得到不饱和酯21/21'的混合物(1:1),再经Dess-Martin氧化及后续自发半缩醛化得到(-)-3-Oxochatancin (4)及其C3区域异构体C1-epi-1(5.2:1,95%),从而确定了其绝对构型。在80 ℃下,用DBU处理4以82%的产率得到( )-Sarcophytin (1)。此外,用RuCl3·3H2O、K2S2O8和KOH原位生成的钌酸钾(K2RuO4)处理21/21'经直接氧化/差向异构化/半缩醛化也可以得到1(68%)。
遗憾的是,未能实现1到(-)-Isosarcophytin (2)的转化:1对热(甲苯,180 ℃,封管)和碱性(DBU, 甲苯, 110 ℃或NaOMe, MeOH, 65 ℃)条件均呈惰性,而在酸性条件(如催化量p-TsOH·H2O, PPTS, TMSOTf等)下迅速脱水得到22。上述结果促使作者重新鉴定了2的原始结构,并发现合成样品4与天然(-)-Isosarcophytin具有相同的NMR数据。因此,作者将(-)-Isosarcophytin的结构修正为4。甲苯磺酰腙23经NaBH3CN/p-TsOH·H2O处理脱除酮羰基得到24(75%)。随后,24在Mn(dpm)3-催化下水合得到反式二醇7及其C10差向异构体(6.5:1,83%)的混合物。按照合成4的路线,将7进一步转化为( )-Chatancin (3)。

(图片来源:Angew. Chem. Int. Ed.)
作者和浙江大学洪鑫课题组合作,通过密度泛函理论(DFT)计算解释了4和C1-epi-1以及1和C1-epi-4之间的热力学平衡(Scheme 4),并且计算的热力学能量证实了实验结果。控制4和C1-epi-1之间平衡的关键因素是共轭酯的几何形状(Scheme 4a)。4的结构具有优势的s-trans几何结构,而相同的片段在C1-epi-1中具有s-cis几何结构,这是由两个羰基之间的空间排斥引起的。脱除酯基后将导致逆向热力学偏向。对于C1-epi-4和1之间的平衡,由于桥碳原子和异丙基之间的空间排斥,导致C1-epi-4不稳定(Scheme 4b)。

(图片来源:Angew. Chem. Int. Ed.)
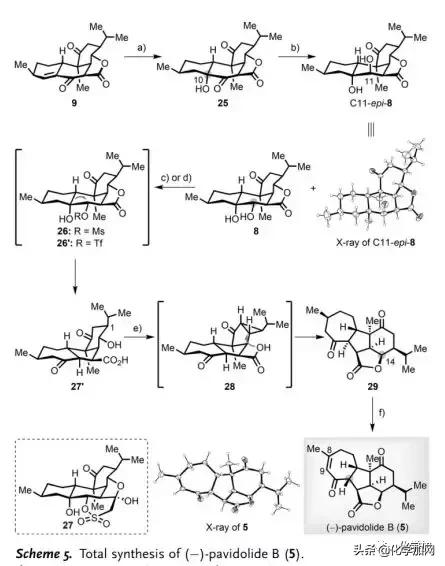
(-)-Pavidolide B(5)的全合成(Scheme 5):9经Mukaiyama水合可以选择性得到α-醇25(76%)。随后,作者筛选了一系列还原剂如Zn(BH4)2、NaBH4/Et3B、L-Selectride、DIBAL-H和SmI2用于C11位酮的区域和非对映选择性还原,并且过度还原会产生三醇的异构体混合物或仅得到反式二醇C11-epi-8(Scheme 5)。幸运的是,作者发现用NaBH4/Py处理25可以得到8为主要异构体(78%),为后续频哪醇重排奠定了基础。然而,在各种Lewis酸条件下,甲磺酸盐26均会发生分解,并且在碱性条件下发生分子内aldol环化产生五环化合物27。令人高兴的是,三氟甲磺酸酯26'不经分离,在t-BuOK/t-BuOH存在下直接转化成预期产物27'(69%)。在构建出[6,5,7]三环母核骨架后,作者将注意力转向内酯关环、C14位立体化学反转、C11位差向异构化以及引入Δ8,9-双键。一方面,利用Mitsunobu反应未能实现27'内酯化;另一方面,由于C1位异丙基的空间位阻影响,利用其甲磺酸盐进行分子内SN2环化得到29的产率很低(<10%)。因此,作者不得不采取另一种策略。经过详尽的试验,作者发现用亚化学计量的BF3·Et2O可以实现27'缩环得到[3,5,5,7]四环化合物28,然后用羧酸进行分子内亲核加成导致环丙烷开环得到29(84%)。最后,利用Mukaiyama报道的脱氢方法,区域选择性构建出Δ8,9-双键,伴随在C11位差向异构化得到(-)-Pavidolide B(5,75%)。
(图片来源:Angew. Chem. Int. Ed.)
小结: 丁寒锋、林旭锋课题组 以易得的醇 (-)- 12 为原料经过 8 ~ 10步 反应完成了四种西松烷型二萜类化合物: ( )- S arcophytin 、 ( )- C hatancin 、 (-)-3- O xo c hatancin 和 (-)- P avidolide B的 全合成,并对 (-)- I sosarcophytin的结构进行了修 正。其合成策略包括双重 Mukaiyama Michael 加成 /消除 、 Helquist 环化、两种底物控制的面选择性水和以及频哪醇重排等 。此外,后期BF 3 ·Et 2 O- 介导的直接内酯化也是其成功的关键。更重要的是,这些化合物的全合成揭示了两种不同多环骨架之间可能的生源关系。
撰稿人:爽爽的朝阳
,免责声明:本文仅代表文章作者的个人观点,与本站无关。其原创性、真实性以及文中陈述文字和内容未经本站证实,对本文以及其中全部或者部分内容文字的真实性、完整性和原创性本站不作任何保证或承诺,请读者仅作参考,并自行核实相关内容。文章投诉邮箱:anhduc.ph@yahoo.com






