生物拖尾效应(M.C.White团队Nature)
本文来自X-MOLNews
在药理学中有一种非常经典的学说——“占领学说(Occupation Theory)”,认为“受体只有与药物结合才能被激活并产生效应”。药物与受体的相互作用除了静电相互作用和立体互补外,构象、去溶剂化以及取代结合位点水分子等都会对亲和力产生影响。其中,甲基扮演了重要角色,被药物化学家称为“神奇的甲基效应(Magic Methyl Effect)”。这也无怪乎甲基虽为最小的烷基片段,却存在于67%的畅销药(Top 200)中。这是因为在先导化合物的优化过程中,特定位点的甲基化可以通过空间位阻作用提高代谢稳定性(如辛伐他汀),或在代谢位点引入甲基缩短半衰期(如依托考昔)。当然,引入甲基还可能改变药物溶解性和对其他靶点的选择性,甚至完全改变药物的效应,例如将激动剂变成拮抗剂。

引入甲基提高活性的例子。图片来源:Nature
尽管甲基广泛存在于小分子药物中,但通常引入甲基的方法是从头合成(de novo synthesis),目前仍缺乏一种通用的甲基化手段实现复杂分子体系的后期修饰(late-stage modification),从而导致合成效率低,也无法在老药上快速做文章。最近十年,合成化学家已经意识到在含氮或含氧杂环的邻近杂原子位点直接进行C(sp3)–H键烷基化才是这一问题的最优解,并且做了大量努力,例如C(sp3)–H键金属化、过渡金属催化的C(sp3)–H键活化或单电子转移反应(SET)。虽然这些方法可以使用多种烷基化试剂,但底物往往是比较简单的氮杂环或具有特定的结构。因此,实用的甲基化方法必须要具有以下特点:底物范围广(如各种杂环)、官能团兼容性好(如可以耐受羰基、氰基等亲电性基团)、手性保持(反应前后原手性中心不发生差向异构化)以及反应彻底(因为甲基是最小的电中性基团,甲基化产物往往与原料难分离)。

常用的C(sp3)–H直接甲基化方法。图片来源:Angew. Chem. Int. Ed., 2013, 52, 2
在这种背景下,合成化学家发现含氮或含氧杂环的邻近杂原子位点可以先进行C(sp3)–H键羟基化,转化为亚胺离子或氧鎓离子中间体再实现甲基化。然而,此前报道的方法都依赖于底物结构来控制选择性,位点选择性和化学选择性均不理想,因此无法应用于复杂体系。同时,由于缩醛胺或半缩醛中间体存在很强的超共轭效应,不可避免地被过度氧化成羰基,导致在甲基化操作前/后引入还原步骤,步骤经济性不高。这个时候,催化剂控制的氧化甲基化策略走进了合成化学家的视野,因为这种方法极有可能适用于更广泛的底物。
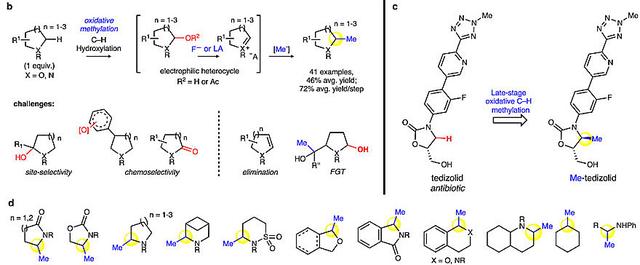
通过氧化甲基化来实现杂环2位C(sp3)–H键甲基化。图片来源:图片来源:Nature
2019年,美国伊利诺伊大学厄巴纳-香槟分校的M. Christina White教授(点击查看介绍)课题组首次利用Mn(CF3PDP)(MeCN)2(SbF6)2(1)实现了芳香烃分子中非活泼亚甲基C(sp3)–H键的高选择性羟基化(Nat. Chem., 2019, 11, 213-221)。该反应的官能团兼容性好,但仅限于缺电子芳烃,电中性、富电子芳烃以及杂芳烃仍是较大挑战。于是,他们设想能否利用这种大位阻的催化剂实现杂环化合物的α-C(sp3)–H键羟基化?近日,他们经过一系列条件筛选,可在不同复杂程度的含氮、含氧杂环化合物上实现α-C(sp3)-H键的选择性甲基化,相关成果发表在Nature上。
首先,研究人员选择芳基化的γ-内酰胺2作为模板底物,在10 mol% 1、5 equiv. H2O2、15 equiv. AcOH下反应,主要得到过度氧化的酰亚胺4b;当降低催化剂和氧化剂用量(0.5 mol% 1, 2 equiv. H2O2)后,主要得到缩醛胺4a。值得注意的是,4a在强力的条件下仍可转化为4b。尽管反应不彻底,但并不需要分离原料和羟基化中间体,因为都可以直接转化为甲基化产物3。经过条件摸索,研究人员发现DAST、Deoxo-Fluor或BF3•OEt2都可以作为羟基活化剂使其转化为亚胺中间体,而廉价易得的AlMe3可作为甲基化试剂,同时还能有效避免烯胺副产物的生成(碱性条件如三乙胺则有利于此过程)。另外,将AlMe3换成格氏试剂产率明显降低,因为后者官能团兼容性较差,不过甲基格氏试剂可用于链状的二级酰胺和碳环的C(sp3)–H键甲基化。

条件筛选。图片来源:图片来源:Nature
在此基础上,研究人员首先在一些简单但在药物中常见的饱和杂环上进行底物扩展(其中活化的Lewis酸需要根据具体情况选择)。在DAST活化后,成功以克级规模实现2的甲基化,收率为71%;也可以用市售AlEt3以51%的收率实现2的乙基化。δ-内酰胺、噁唑烷酮类化合物也能兼容该反应,以中等的收率得到目标产物(6-8)。对于药物中最常见的氮杂环——吡咯烷,能以54%的收率得到单甲基化产物9;对于高活性取代基(如3° 叔碳、3° 苄基、3° α-羰基)取代的底物,均在位阻较小的亚甲基位点上发生甲基化(10-13)且可以手性保持(12、13);对于3-苯基吡咯烷衍生物,在远离苯基的亚甲基上发生甲基化生成5-甲基化产物16;此外,甲酯、酮、乙酸盐、腈也能够耐受该反应(12-15)。对于小分子药物中常见的氮杂环——哌啶类化合物以及其它的环状胺(如氮杂环庚烷、十氢喹啉等)也能以中等的收率得到目标产物(17-23)。另外,氧杂环化合物也能以74%的收率实现甲基化(26)。

底物扩展。图片来源:Nature
前文已经提到,甲基化操作有时会提高分子的生物活性或选择性,从而提高这些化合物的靶向性、安全性甚至生理效应,称为“神奇的甲基效应”。在上述简单的杂环上取得成功后,研究人员将目光投向结构更复杂的药物分子上。结果显示23个含有不同杂环的药物分子或其前体都能在特定位置上进行选择性甲基化(27-50,活化的Lewis酸需要根据具体情况选择)。例如钾通道活化剂醋酸克罗卡林(27)、脊髓性肌萎缩症抗炎药吲哚洛芬甲酯(29)、镇咳药芬斯匹利(31)、抗抑郁药西酞普兰(36)都能以中等的收率实现氧化甲基化;对于抗抑郁药diclofensine衍生物,尽管存在极富电子的甲氧基苯基,仍能在四氢异喹啉的亚甲基上实现甲基化生成34。吡咯并异喹啉前体(具有神经递质摄取抑制剂特性)在空间位阻较小的亚甲基上发生甲基化,以44%的收率得到37,而不是活性更高的叔基、苄基。值得一提的是,抗炎药塞来昔布的哌啶衍生物能够以良好的收率得到单甲基化产物41。基于脯氨酸的二肽、三肽、四肽均能以较好的收率实现甲基化(42-44);天然萜类化合物Ambroxide也能以良好的收率在四氢呋喃环的亚甲基上实现甲基化(45)。值得注意的是,RORc反向激动剂46的高级中间体可在1/H2O2/TMSOTf/AlMe3下实现氧化甲基化,得到顺式非对映异构体47,随后在钯催化下进行交叉偶联得到( )-Me-RORc反向激动剂,而先前的方法需要六步才能实现其合成,总产率仅为1.4%。
最后,研究人员还考察了杂环之外的底物,发现氧化C-H键甲基化并不局限于可以形成亚胺离子或氧鎓离子中间体的底物,例如链状的苄胺也能实现甲基化,得到S1P1拮抗活性提升2135倍的前药52;但是需要亲核性更强的甲基溴化镁试剂。有意思的是,该方法还能在阿比特龙类似物53的饱和碳环上进行选择性甲基化,这也是首次报道的非活化C(sp3)–H键甲基化反应。同时也体现出该方法对药物分子的后期修饰具有很高的步骤经济性,可避免从头合成的麻烦,从而加速药物的发现和优化。

在生物活性分子后期修饰中的应用。图片来源:Nature
总结
无数药物发现实例证明,小小甲基就像有“魔法”一样可以改变药物分子的性质。但这个“魔法”并不容易施展,长期以来C(sp3)–H键甲基化仅仅停留在对简单底物的操作上,缺乏更广泛的底物范围,难以在复杂分子上应用。M. C. White团队此项工作无疑提供了一种更强大的“魔杖”,期待药物化学家能用它更快发现更多良药。
Late-stage oxidative C(sp3)–H methylation
Kaibo Feng, Raundi E. Quevedo, Jeffrey T. Kohrt, Martins S. Oderinde, Usa Reilly, M. Christina White
Nature, 2020, DOI: 10.1038/s41586-020-2137-8
导师介绍
M. Christina White
https://www.x-mol.com/university/faculty/212
(本文由峰千朵供稿)
,免责声明:本文仅代表文章作者的个人观点,与本站无关。其原创性、真实性以及文中陈述文字和内容未经本站证实,对本文以及其中全部或者部分内容文字的真实性、完整性和原创性本站不作任何保证或承诺,请读者仅作参考,并自行核实相关内容。文章投诉邮箱:anhduc.ph@yahoo.com






