可膨胀石墨ph测试(埃米级精准调控氧化石墨层间距)

第一作者:Jiyoung Lee, Chanhoon Kim
通讯作者:Il-Doo Kim
通讯单位:韩国科学技术研究院(KAIST)
【研究背景】
随着人们对气候变化和能源安全的日益关注,电动汽车引起了人们的广泛关注,并具有广阔的市场前景。然而,电动汽车的实际应用仍然受到当前LIBs长充电时间的阻碍。虽然可以提高功率减少充电时间,但随着LIBs充电速率的增加,会在石墨负极上电镀金属锂,这不仅会导致电池容量的严重衰减,还可能增加短路的风险。为了提高对电动汽车的需求,设计支持快速充电的可充电电池至关重要。然而,商用LIBs由于石墨负极狭窄的层间间距导致缓慢嵌入的动力学,倍率性能受限。因此调节石墨材料的夹层距离,通过缓解扩散阻碍来提高电化学动力学,实现可以快速充电的负极材料。
【成果简介】
鉴于此,韩国科学技术研究院(KAIST)Il-Doo Kim教授(通讯作者)提出了一种通过简单的溶剂热反应在Å水平上进行层间调制的超精密策略,通过使用三种α, ω-二氨基功能化的有机填料,成功地合成了具有7.4-13 Å之间精细控制的氧化石墨框架(GOF)。不仅有机填料的二元胺官能团与GO表面氧化基团形成共价交联,同时还化学还原GO层。最后获得了具有改善的电子特性和扩展的GOFs,表现了前所未有的倍率性能(在3000 mA g-1电流下370 mAh g-1容量,对应5 min充电)。相关研究成果以“An angstrom-level d-spacing control of graphite oxide using organofillers for high-rate lithium storage”为题发表在Chem上。
【核心内容】
如图1A所示,氧化石墨框架(GOFs)通过溶剂热法分别与1, 4-苯二胺(P)、2, 6-二氨基蒽醌(DQ)和乙二胺(E)有机填料反应合成了P-GOF、DQ-GOF和E-GOF。每种填料分别具有亚甲基链分子、一个苯环和三个苯环,导致了层间间距的差异,而且各种官能团(如环氧,羟基,羰基和羧基)提供了活性位点,有效地加宽了GO层间距。GOF的XRD图谱如图1B,E-GOF、DQ-GOF和P-GOF的层距离分别为7.4、9.8到13.0 Å,呈现可调且增加的趋势,而纯的GO大约为9.0 Å。最长的填料插入的DQ-GOF显示出比P-GOF更小的层隙,这归因于DQ填料大的位阻、有限的灵活性和强的π-π堆积,表明填料链(DQ)可以平放放置。如图1C为FT-IR图谱,插入填料后,可以清楚地观察到宽的OH伸缩振动(3200-3400 cm-1)的消失和C=N伸缩振动(1650 cm-1)和C–N伸缩振动的出现(1305 cm-1对于OC–N键、1080 cm-1对于C-N键),从而表明GO层之间填料单元的成功交联。在DQ-GOF红外光谱中出现轻微的峰值在1730 cm-1,对应于DQ官能团的C=O伸缩振动。通过XPS图谱(如图1D)与原始GO相比,GOF具有增加量的N和减少量的O原子,表明填充物通过GO表面上氨基的交联在GO片层之间起协调作用。C=C键的显著增加以及C-N键的出现进一步证实了含氮部分取代了氧官能团,GOFs丰富的表面官能团有助于与Li 的良好相互作用。
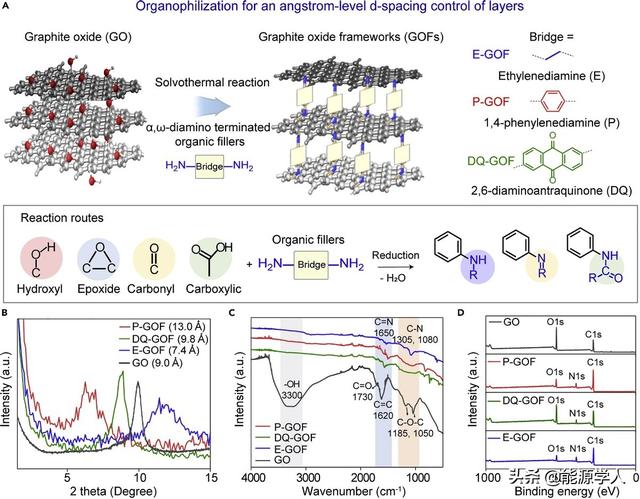
图1. GOFs的合成和表征:(A)三种不同的有机填料的GOFs合成示意图, (B) XRD图谱,(C)傅里叶变换红外光谱,(D)XPS图谱。
为了证明扩大层间距的影响,对GOFs进行了长循环性能。如图2A所示,所有的GOFs在高电流密度下保持了优异的锂储存性能,P-GOF、DQ-GOF和E-GOF在3000 mA g-1电流密度下循环了1500次仍保持了280、180和370 mAh g-1的容量,而石墨由于其较低的层间距(3.45 Å),仅呈现10 mAh g-1非常低的容量。与之前的报道相比,E-GOF在充电速度和容量方面表现出相当的电化学性能(如图2B)。此外,随着循环增加容量的持续增加,可归因于电极表面上的赝电容型聚合物膜,导致活性材料内部反应位点的缓慢活化,从而诱导Li 在表面和面内附近逐渐吸附。除了容量的提高,GOFs还表现出优异的倍率性能,归因于层间间距的扩大。在100-5000 mA g-1的电流密度范围内,所有的GOFs都表现出比石墨高得多的倍率容量和可逆容量(如图2C)。总的来说GOFs具有高的锂存储性能,因此膨胀层状结构的GOFs具有作为快速充电和高容量负极材料的特征。

图2. GOFs系列电极的电化学性能:(A)在3000 mA g-1电流密度下的长循环稳定性,(B)GOFs和先前报道的石墨烯/石墨衍生物材料之间能量储存性能的比较,(C)不同电流密度下GOFs的倍率性能。
为了深入了解层间距离对Li 扩散的影响,如图3A-C进行EIS分析。在高频区和低频区存在两个半圆,分别对应SEI膜的阻抗(RSEI)和电极/电解液的界面电阻(RCT),在低频区存在一条直线,对应于在活性材料中的扩散阻抗(Zw)。根据30 ℃下的EIS,随着层间距离的增加,相应的RSEI具有更高的电阻值。结果表明,由于电解质的过多侵入,在扩展的层间间隙内形成了较厚的SEI膜。SEI膜的厚度通过XPS深度剖面进一步测量(如图3D),在284.5eV处的图谱表明,与DQ-GOF和P-GOF相比,E-GOF在电极表面形成了更薄的SEI,这与EIS分析中的观察结果一致。基于图3A-C的EIS图谱计算活化能(Ea),可以分为ESEI和ECT。相比于先前的报告石墨负极活化能(60-70 kJ mol-1),合成的GOFs具有更低的Ea,其中E-GOF呈现低于1.3-1.5倍的ESEI和ECT值,表明在电极/电解液界面形成的SEI膜薄且稳定(如图3E)。这也可能归因于GOFs扩展的层间间距导致低接触电阻,有利于电子传输通过界面,而P-GOF和DQ-GOF都显示出显著增加的ESEI,归因于电解质过多的侵入,过多的SEI膜也会增加ECT。结果表明E-GOF表现出较低的Ea,提高了电池的电化学性能。如图3F在不同的锂化状态下进行了恒电流间歇滴定技术(GITT),随着放电深度的增加所有GOFs的DLi都下降,这可归因于在低电位状态下动力学限制固态扩散贡献。最重要的是,E-GOF显示最高的DLi值,坡度平缓,沿下降趋势没有大的下降,而P-GOF和DQ-GOF表现出较陡的坡度,变化较大。这表明具有适当膨胀的层状结构的E-GOF在离子扩散性方面具有最好的电化学性能。

图3. GOFs电极的电荷传输行为:(A-C)在30-80 ℃不同温度下测试循环100圈后的EIS,(D)C1s光谱的XPS深度分布图,(E)活化能量的比较,(F)通过GITT分析GOFs电极的锂离子扩散系数,并作放电深度(DOD)的函数。
如图4A-C为从0.1到5 mV s-1的不同扫描速率下CV曲线,通过分析计算阐明GOF电极内的离子存储机制,包括电容控制反应(b值接近1)和扩散控制反应(b值接近0.5)。通过计算可以得到所有的峰C2的b值接近0.5,而E-GOF、P-GOF和DQ-GOF峰C1的b值分别为0.93、0.95和0.78(如图4D-F),分别对应于扩散控制和电容控制为主。进一步计算如图4G-I,在1 mV s-1扫描速率下P-GOF、DQ-GOF和E-GOF的电容控制反应贡献的百分比分别为45%、37%和40%。结果表明具有最大层间距的P-GOF具有最高比例的电容反应,而与DQ-GOF的情况相比,E-GOF对电容反应的贡献更大,这表明DQ填料单元可能会干扰离子渗透路径。

图4. GOFs电极的电荷存储机制:(A-C)不同扫描速度下的CV曲线,(D-F)在特定峰电流下log(i)和log(v)的函数关系,(G-I)在1 mV s-1扫速下电容行为所占的百分比。
基于上述结果,与石墨相比,GOFs电化学性能的提高直接归因于充分扩大的层间距,如图5所示。GOFs增大的层间间距允许Li 容易插入片层之间,并降低Li 运输过程的能量势垒,导致快速扩散动力学。此外,由于GOFs的层间距增大, GO和二氨基的有机填料结构的缺陷和表面官能团暴露出来,为Li 提供了额外的电活性位点和表面积。更重要的是,发现GOFs中的快速离子传输动力学是由电容行为、大的层间距以及层内氧化物官能团的消除引起的,导致在高电流密度下优异的锂存储特性。然而,过高的电流密度可能会引起较大的ESEI以及在GOFs的表面上形成厚的SEI,特别是在它们的夹层内部形成过量的SEI层。

图5. 层间结构与性能的关系,不同层间距下锂离子的扩散行为和储能性能示意图。
【结论展望】
本文提出了一种简单而超精密的方法来控制GOs的层间间距,使用有机填料来制备高性能负极材料。依靠在二元胺有机填料的官能团和GO表面氧化物基团之间形成共价键,成功地合成了一种有机框架结构,根据填料的选择控制层间距在7.4到13 Å的范围内。此外,通过三种填料确定了GOFs的最优层间距,其以最小的势垒进行Li 的运输。优化的GOF电极显示出特别快的充电速率和高锂储存容量。不仅为设计快速充电锂离子电池提供了重要的见解,而且促进了层状结构材料的合理开发。
【文献信息】
Jiyoung Lee, Chanhoon Kim, Jun Young Cheong, and Il-Doo Kim*, An angstrom-level d-spacing control of graphite oxide using organofillers for high-rate lithium storage, 2022, Chem https://www.sciencedirect.com/science/article/pii/S245192942200225X
,免责声明:本文仅代表文章作者的个人观点,与本站无关。其原创性、真实性以及文中陈述文字和内容未经本站证实,对本文以及其中全部或者部分内容文字的真实性、完整性和原创性本站不作任何保证或承诺,请读者仅作参考,并自行核实相关内容。文章投诉邮箱:anhduc.ph@yahoo.com






